Do, 22.03.2018 - 06:04 — Redaktion 
![]()
Das Ableben des weltberühmten Physikers Steve Hawking hat uns die Problematik von neurodegenerativen Erkrankungen wieder vor Augen geführt. Für die Amyotrophe Lateralsklerose, an der er litt, gibt es nach wie vor keine Therapie und viele Versuche das Absterben der Nervenzellen zu verlangsamen, haben fehlgeschlagen. Eine Studie, die eben im open access Journal eLife erschienen ist, zeigt einen neuen Ansatz, der darin besteht, dass dié initiale Degeneration der Nerven -Muskel -Verbindung (= Synapse) verhindert wird [1]. Ein erfolgreicher Schutz der Synapsen könnte auch als Therapie in anderen neurodegenerativen Erkrankungen zur Anwendung kommen. Der Neuropatholologe und ALS-Experte Jonathan D. Glass (Emory University ALS Center, Atlanta) hat die Ergebnisse dieser Studie zusammengefasst [2].*
In der Neurobiologie gilt ganz allgemein, dass die Intaktheit eines Axons - es handelt sich dabei um die kabelähnlichen Fortsätze, die Informationen zwischen Nervenzellen (Neuronen), Muskeln und sensorischen Rezeptoren übertragen - völlig davon abhängt, wie gesund der Zellkörper des Neurons ist. Diese Vorstellung war daher auch Leitprinzip in der Suche nach experimentellen Therapeutika zur Behandlung von neurodegenerativen Erkrankungen, beispielsweise von Alzheimer, Parkinson und amyotropher Lateralsklerose. Der Schutz des Zellkörpers des Neurons sollte dazu beitragen, das Voranschreiten der Neurodegeneration zu verlangsamen oder sogar zu stoppen. Wie ein Neuron aussieht, ist in Abbildung 1 schematisch dargestellt.
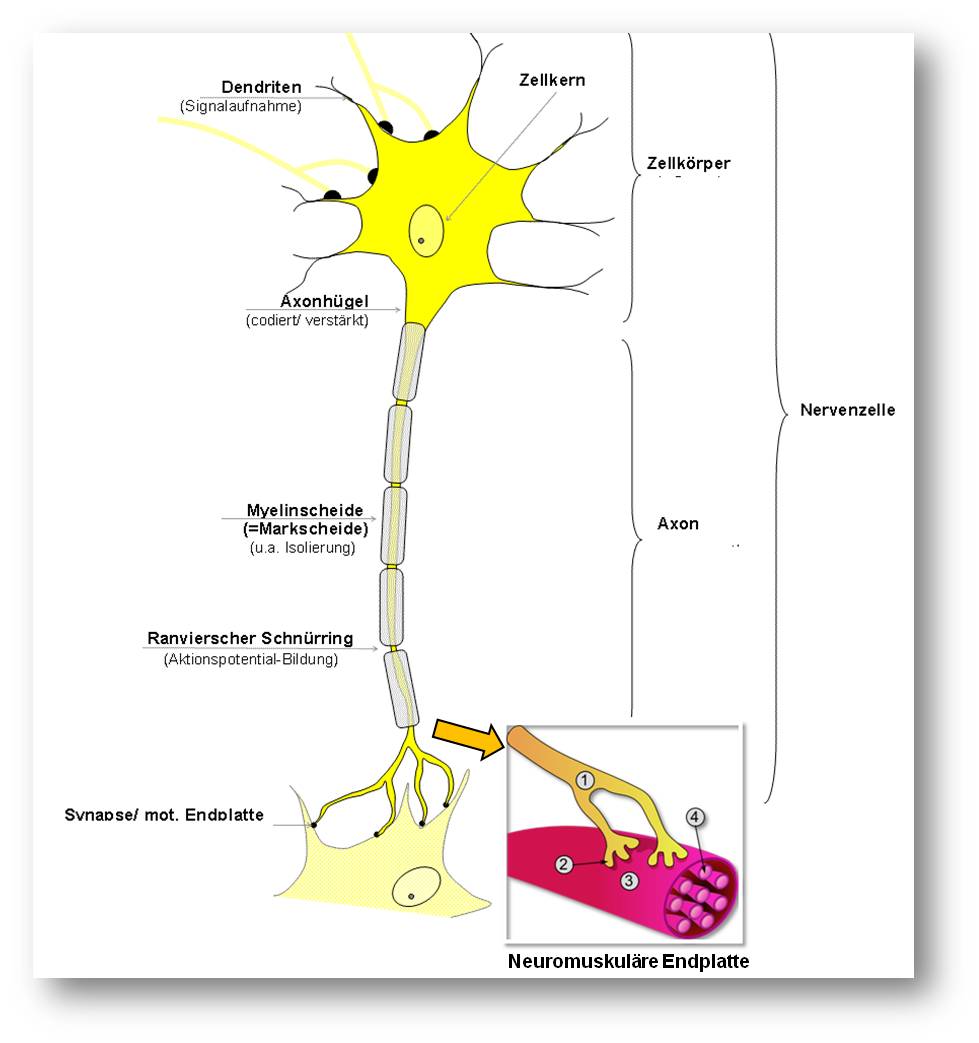
.Insert rechts unten: Neuromuskuläre Endplatte. 1: terminales Axon eines Motoneurons, 2: neuromuskuläre Endplatte, 3: Muskelfaser, 4: Myofibrille. (Bild von der Redaktion eingefügt; Quelle: fr:Utilisateur:Dake, https://de.wikipedia.org/wiki/Nervenzelle#/media/File:Synapse_diag3.png; Lizenz: cc-by-sa 3.0)
Aus Tierexperimenten und Beobachtungen am Menschen kommen allerdings mehr und mehr Hinweise, dass Neuronen, Axone und auch Nervenendigungen in unterschiedlicher Weise auf Schädigungen reagieren. Dies legt den Schluss nahe, dass unabhängige Mechanismen der Neurodegeneration in verschiedenen Abschnitten des Neurons eine Rolle spielen. Um eine wesentliche therapeutische Wirkung zu erzielen, müssten Behandlungen daher auf das Neuron als Ganzes abzielen.
Amyotrophe Lateralsklerose (ALS)
ist eine verheerende Erkrankung der Motoneuronen, die ohne Vorwarnung, typischerweise in der Blüte des Lebens auftritt. Menschen mit ALS werden zunehmend schwächer -es folgt ein grausamer Weg, der über Behinderung zum Verlust der Selbständigkeit und schlussendlich zum Tod führt. Therapeutische Maßnahmen sind weitgehend palliativ - Dutzende klinische Studien haben in den letzten drei Jahrzehnten erfolglos nach Mitteln gesucht, die ein Fortschreiten dieser Krankheit zumindest verlangsamen sollten.
Präklinische Experimente an ALS- Krankheitsmodellen in Tieren haben zwar vielversprechende Ergebnisse gezeitigt, konnten aber leider nicht erfolgreich auf den Menschen übertragen werden; warum diese Fehlschläge passierten,wird viel diskutiert. Besonders interessant - vor allem, weil die Ergebnisse paradox sind - waren Tierversuche, die zwar einen nahezu vollständigen Schutz der Zellkörper von Motoneuronen aufzeigten, jedoch das Voranschreiten der Krankheit nicht verzögern konnten: Es wurden dabei verschiedenartige Techniken angewandt, um die Zellkörper der Motorneuronen zu schützen; der Verlust der Verbindung (Konnektivität) zwischen den Nervenendigungen und dem Muskel (die sogenannte Denervierung) konnte damit aber nicht verhindert werden.
Der degenerative Prozess…
Experimentelle Untersuchungen an ALS- Tiermodellen haben gezeigt, dass der degenerative Prozess (als Absterben ("dying-back") bezeichnet) an den Nervenendigungen beginnt und das Axon entlang bis schließlich hin zum Zellkörper des Neurons fortschreitet. Wie bei jedem "elektrischen" System führt die Durchtrennung von Draht (Axon) und Zielobjekt (dem Muskel) dann zum Verlust der Funktion - im Fall von ALS sind Schwäche und schliesslich der Tod die Konsequenz.
Der Prozess beginnt also dort, wo das Motoneuron an den Muskel bindet, an der sogenannten neuromuskulären Endplatte (neuromuscular junction; siehe Abbildung 1), einer speziellen Synapse, an der chemische Signale (der Neurotransmitter ist Acetylcholin; Anm. Redn) zur Aktivierung der Muskelkontraktion übertragen werden.
…und eine mögliche Gegenstrategie
Im Journal eLife berichten nun Wissenschafter von der NYU Medical School und der Columbia University, dass ein Erhalt der Verbindung zwischen Nerv und Muskeln den Krankheitsverlauf verlangsamen könnte [2].
Um diese Nerv-Muskel-Verbindung aufrecht zu erhalten, sind zahlreiche Proteine involviert, u.a. ein Rezeptor-Protein namens MuSK. Dieses MuSK stimuliert während der Entwicklungsphase die Anheftung der Nervenendigung an den Muskel. Später im Leben ist MuSK notwendig, um die neuromuskuläre Endplatte stabil zu erhalten und damit zu gewährleisten, dass Signale zwischen Muskel, Nervenendigung und motorischem Axon laufen können. Tatsächlich sind Schädigungen oder Mutationen des MuSK-Gens mit neuromuskulären Erkrankungen verbunden. Abbildung 2.
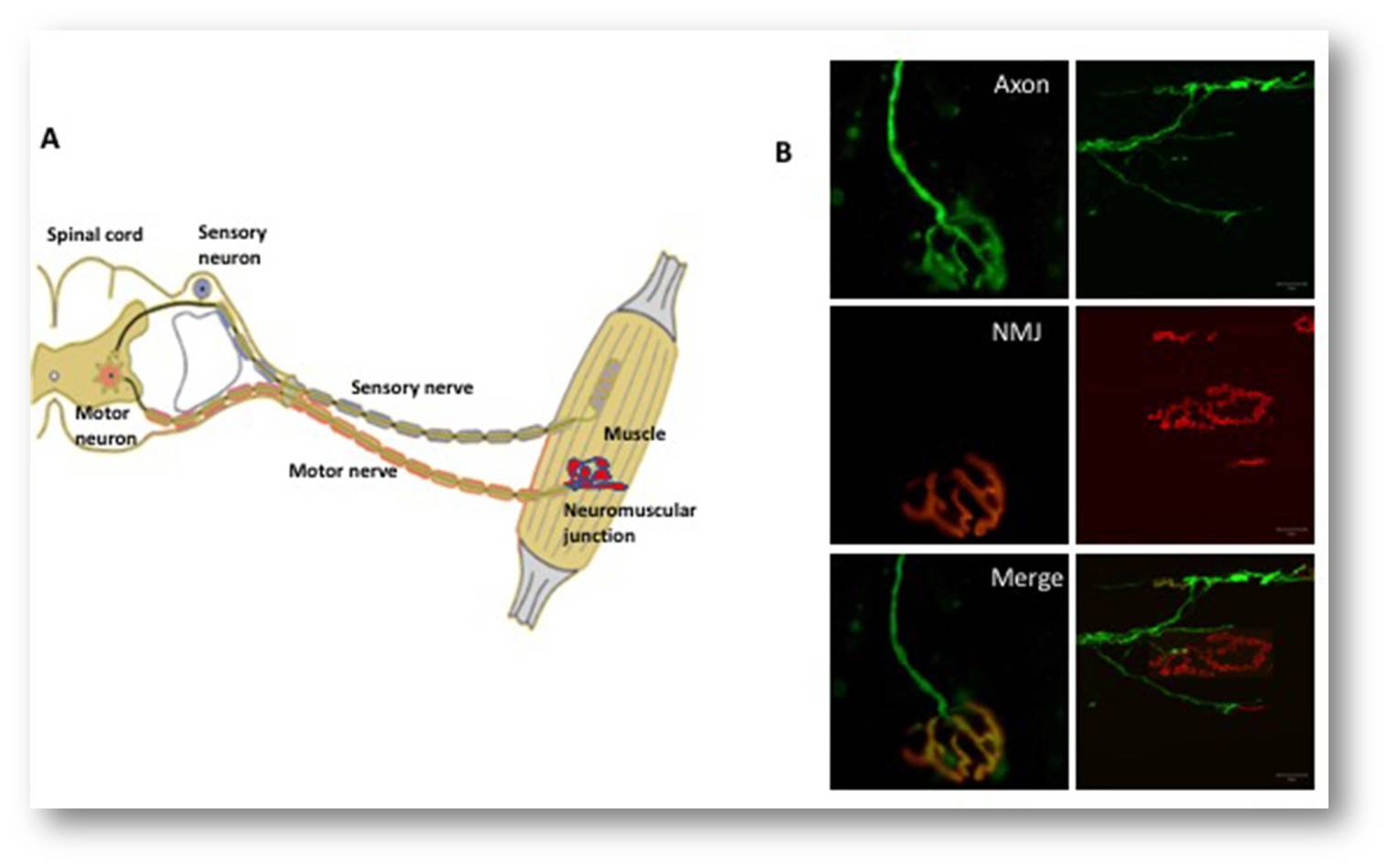 Abbildung 2. Das somatische Nervensystem. (A) Eine schematische Darstellung des neuromuskulären Systems. Motoneurone (rot) im Gehirn und im Rückenmark senden elektrische Impulse entlang der Axone (die den motorischen Nerv bilden) zu den Muskelfasern, um sie zu kontrahieren. Die sensorischen Neuronen (blau dargestellt) übermitteln Informationen (in Form von elektrischen Impulsen) von verschiedenen Körperteilen an das Gehirn. Bei Erkrankungen wie der amyotrophen Lateralsklerose gibt es Hinweise darauf, dass die Degeneration an der neuromuskulären Endplatte zwischen den Enden der Axone und dem Muskel beginnt und entlang des Axons in Richtung des Zellkörpers fortschreitet. (B) Fluoreszenzmikroskopie der neuromuskulären Endplatte (neuromuscular junction - NMJ). Die Felder auf der linken Seite zeigen eine vollständig innervierte neuromuskuläre Endplatte, wobei das Axon grün (oben links) und der Muskel rot markiert ist (Mitte links). Das zusammengefügte Feld (unten links) zeigt die vollständige Überlappung des Nervenendstücks mit dem Muskel (gelb). Die Platten rechts zeigen eine denervierte neuromuskuläre Endplatte, in der das Axon nicht mehr mit dem Muskel überlappt.
Abbildung 2. Das somatische Nervensystem. (A) Eine schematische Darstellung des neuromuskulären Systems. Motoneurone (rot) im Gehirn und im Rückenmark senden elektrische Impulse entlang der Axone (die den motorischen Nerv bilden) zu den Muskelfasern, um sie zu kontrahieren. Die sensorischen Neuronen (blau dargestellt) übermitteln Informationen (in Form von elektrischen Impulsen) von verschiedenen Körperteilen an das Gehirn. Bei Erkrankungen wie der amyotrophen Lateralsklerose gibt es Hinweise darauf, dass die Degeneration an der neuromuskulären Endplatte zwischen den Enden der Axone und dem Muskel beginnt und entlang des Axons in Richtung des Zellkörpers fortschreitet. (B) Fluoreszenzmikroskopie der neuromuskulären Endplatte (neuromuscular junction - NMJ). Die Felder auf der linken Seite zeigen eine vollständig innervierte neuromuskuläre Endplatte, wobei das Axon grün (oben links) und der Muskel rot markiert ist (Mitte links). Das zusammengefügte Feld (unten links) zeigt die vollständige Überlappung des Nervenendstücks mit dem Muskel (gelb). Die Platten rechts zeigen eine denervierte neuromuskuläre Endplatte, in der das Axon nicht mehr mit dem Muskel überlappt.
Das Forscherteam hat nun ein gut charakterisiertes ALS -Modell in der Maus verwendet, um zu testen, ob ein Eingriff bei bereits geschädigten neuromuskulären Synapsen das Fortschreiten der Krankheit verlangsamen kann [2]. Dieser Eingriff bestand darin einen Antikörper zu verwenden, der als Agonist an das MuSK-Protein bindet und dessen Aktivierung stimuliert. Es zeigte sich, dass mit diesem Antikörper die neuromuskulären Synapsen über einen längeren Zeitraum intakt und funktionsfähig blieben. Außerdem trat das Absterben von Motoneuronen verzögert ein - ein Hinweis darauf, dass die Aufrechterhaltung der Innervation des Muskels für das Weiterbestehen des Zellkörpers von Motoneuronen wichtig ist.
Insgesamt überlebten die behandelten Tiere aber nur einige Tage länger als die Kontrollen. Dies könnte auf die aggressive Form der ALS-Erkrankung in diesem Tiermodell zurückzuführen sein oder vielleicht auch darauf, dass die Zellkörper der Motoneuronen nicht auch therapeutisches Ziel waren. Nichtsdestoweniger lassen diese Ergebnisse aber darauf schließen, dass der Schutz der neuromuskulären Synapse eine Möglichkeit darstellen könnte, das Fortschreiten der ALS-Krankheit zu verlangsamen und Neuronen länger am Leben zu erhalten.
Fazit
Pathogenese und Progression der Amyotrophen Lateralsklerose liegen im Dunkeln, sind ein ungelöstes neurobiologisches Problem. Laufend werden Gene und zelluläre Signalwege entdeckt, die mögliche Treiber der Krankheit darstellen und jeweils gezielt manipuliert werden könnten. Die in [2] beschriebenen Ergebnisse lassen einen neuen Ansatz als realisierbar erscheinen, der - auf Basis eines konkreten molekularen Mechanismus - die Bildung und Aufrechterhaltung der Verbindungen zwischen Nervenzellen und Muskeln zum Ziel hat. Damit könnte das Fortschreiten der amyotrophen Lateralsklerose verlangsamt werden. Da ein derartiger Mechanismus aber nicht spezifisch für ALS ist, könnte er auch in anderen neuromuskulären Erkrankungen therapeutische Anwendung finden, bei denen die Denervierung ein wichtiges pathologisches Merkmal ist.
[1] S. Cantor et al., Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody (2018), eLife 2018;7:e34375 doi: 10.7554/eLife.34375 [2] JD Glass, Neuromuscular Disease: Protecting the nerve terminals (Insight Mar 19, 2018). eLife 2018;7:e35664 doi: 10.7554/eLife.35664
*Der von Jonathan D. Glass stammende Artikel: " Neuromuscular Disease: Protecting the nerve terminals" ist am 19. März 2018 erschienen in: eLife 2018;7:e35664 doi: 10.7554/eLife.35664 als eine leicht verständliche Zusammenfassung ("Insight") der Untersuchung von S.Cantor et al. [1]. Der Artikel wurde von der Redaktion ins Deutsche übersetzt und geringfügig für ScienceBlog.at adaptiert (Untertitel, Abbildung 1 aus Wikipedia). eLife ist ein open access Journal, alle Inhalte stehen unter einer cc-by Lizenz -
Weiterführende Links
Homepage eLife: https://elifesciences.org/
Homepage von Jonathan Glass, Prof. und Direktor des Emory ALs Center http://neurology.emory.edu/faculty/neuromuscular/glass_jonathan.html
Emory ALS Center. Video 7:49 min.(englisch) https://www.youtube.com/watch?v=R7PbxcZBvi8 , Standard-YouTube-Lizenz
Infofilm Was ist ALS. Video 2:23 min. https://www.youtube.com/watch?v=IRQb4lkGeVE , Standard-YouTube-Lizenz
Stephen Hawking - Ein persönlicher Nachruf | Harald Lesch (14.3.2018). Video 6:50 min. https://www.youtube.com/watch?v=c1pQS1quRp8. Standard-YouTube-Lizenz
Artikel im ScienceBlog:
Redaktion, 2.04.2017: Wissenschaftskommunikation: das open-access Journal eLife fasst Forschungsergebnisse für die Öffentlichkeit verständlich zusammen. http://scienceblog.at/wissenschaftskommunikation-open-access-journal-elife.
- Printer-friendly version
- Log in to post comments