Do, 03.11.2016 - 07:26 — Richard Neher

![]() Wir sind umgeben von Mikroorganismen, die sich im Wettstreit ums Überleben ständig verändern. Im Unterschied zu Tieren und Pflanzen dauern solche Veränderungen nicht Tausende von Jahren, sondern oft nur einige Wochen. Um solch schnelle Evolution zu verstehen, benötigen wir neue theoretische Konzepte und müssen die evolutionäre Dynamik direkt beobachten. Die Forschungsgruppe rund um den Biophysiker Richard Neher (Arbeitsgruppenleiter am Max-Planck-Institut für Entwicklungsbiologie, Tübingen) entwickelt dazu Methoden und wendet sie auf Daten von Grippe- und HI-Viren an. Die Ergebnisse ermöglichen Vorhersagen der Zusammensetzung zukünftiger Viruspopulationen.
Wir sind umgeben von Mikroorganismen, die sich im Wettstreit ums Überleben ständig verändern. Im Unterschied zu Tieren und Pflanzen dauern solche Veränderungen nicht Tausende von Jahren, sondern oft nur einige Wochen. Um solch schnelle Evolution zu verstehen, benötigen wir neue theoretische Konzepte und müssen die evolutionäre Dynamik direkt beobachten. Die Forschungsgruppe rund um den Biophysiker Richard Neher (Arbeitsgruppenleiter am Max-Planck-Institut für Entwicklungsbiologie, Tübingen) entwickelt dazu Methoden und wendet sie auf Daten von Grippe- und HI-Viren an. Die Ergebnisse ermöglichen Vorhersagen der Zusammensetzung zukünftiger Viruspopulationen.
Virus-Evolution
Evolution gilt gemeinhin als ein langsamer Prozess, der nicht über einen Zeitraum von ein paar Jahren beobachtbar ist. Mikroorganismen hingegen verändern sich oft sehr schnell und können innerhalb von Wochen neue Eigenschaften entwickeln. Zu diesen Mikroorganismen gehören viele Krankheitserreger und deren stetige Anpassung kann dramatische Auswirkungen auf die Gesundheit von Menschen, Tieren und Pflanzen haben, besonders dann, wenn Erreger resistent gegen Medikamente werden.
Die rasante Entwicklung der Sequenzierungstechnologien ermöglicht es, die Veränderung und Ausbreitung von Krankheitserregern, wie zum Beispiel von Grippe-Viren, genau zu beobachten. Das Global Influenza Surveillance and Response System sequenziert jeden Monat Hunderte von Influenza-Viren. Um diese Daten intuitiv und in Echtzeit darzustellen und zu analysieren, hat die Forschungsgruppe um Richard Neher zusammen mit Wissenschaftlern aus Seattle die Webseite nextflu.org (Neher, R.A.; Bedford, T. nextflu: Real-time tracking of seasonal influenza virus evolution in humans, Bioinformatics 31, 3546-3548 (2015)) entwickelt. Abbildung 1 zeigt, wie die Webseite die zeitliche und geographische Ausbreitung von Grippe-Virus Varianten darstellt. Diese Art der Datenanalyse hilft, dass Impfstoffe rechtzeitig an sich verändernde Viren angepasst werden können. Bei anderen Virus Erkrankungen, wie zum Beispiel Ebola, können so Infektionsketten schnell identifiziert werden.
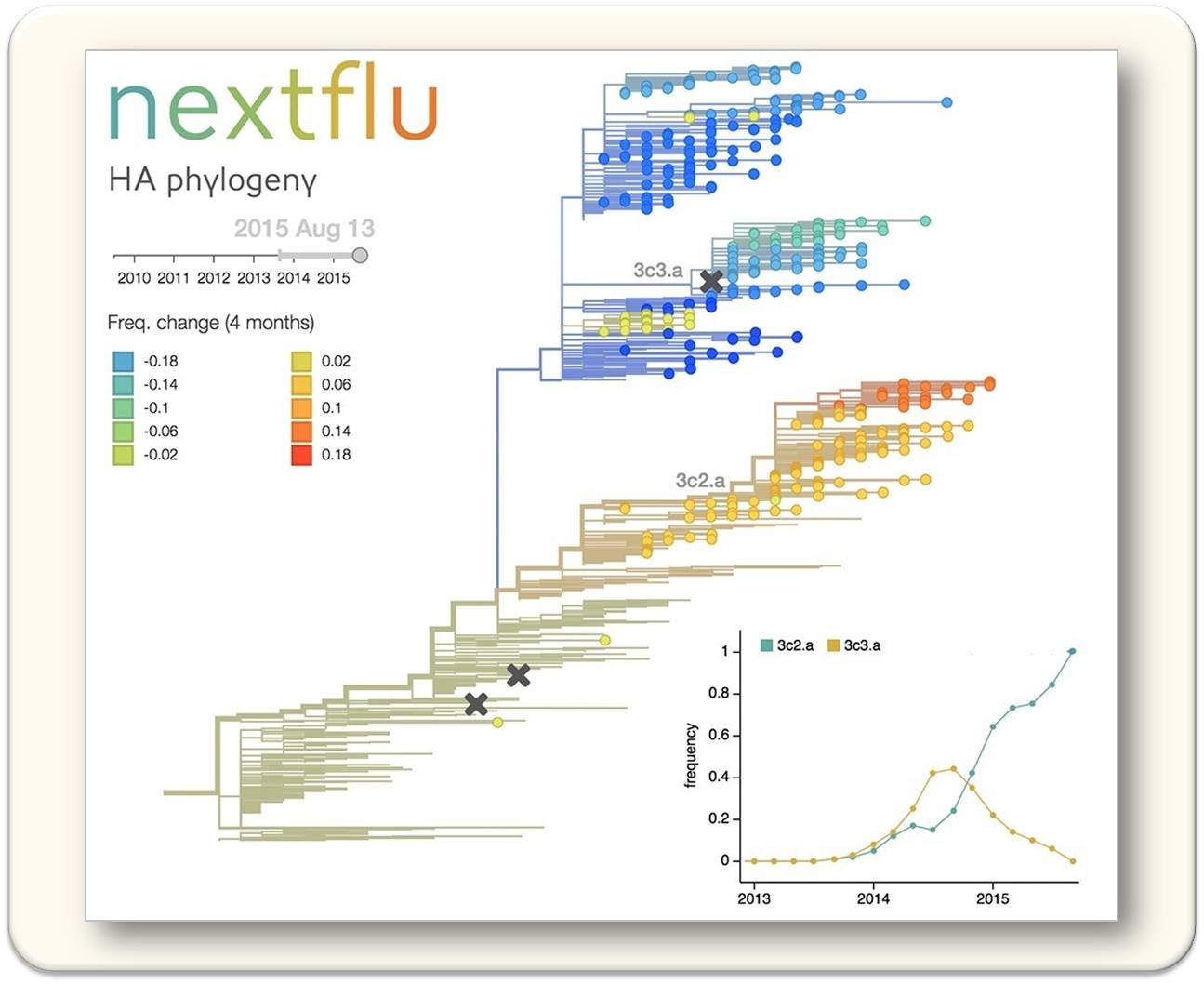 Abb. 1: Fortlaufend aktualisierter Stammbaum und Dynamik von Grippe-Viren, dargestellt auf nextflu.org. Unten rechts ist die Häufigkeit der Virusvarianten 3c2.a und 3c3.a über die Jahre 2013 bis 2015 dargestellt. © Max-Planck-Institut für Entwicklungsbiologie/Neher, Bedford
Abb. 1: Fortlaufend aktualisierter Stammbaum und Dynamik von Grippe-Viren, dargestellt auf nextflu.org. Unten rechts ist die Häufigkeit der Virusvarianten 3c2.a und 3c3.a über die Jahre 2013 bis 2015 dargestellt. © Max-Planck-Institut für Entwicklungsbiologie/Neher, Bedford
Neben der Überwachung der Evolution von Krankheitserregern können wir an den sich schnell verändernden Mikroorganismen evolutionäre Prozesse studieren, die in Eukaryonten Millionen von Jahren dauern würden. Die Fülle an Sequenzdaten aus mikrobiellen Populationen erlaubt Einblicke in die Triebkräfte und Gesetze der Evolution. Deren Verständnis wiederum hilft uns, Bedingungen zu definieren, die die Anpassung von Krankheitserregern verlangsamen. Auch Prognosen der Zusammensetzung und Eigenschaften zukünftiger Viruspopulationen rücken in greifbare Nähe. Diese Entwicklungen spielen sich an der Schnittstelle zwischen neuen Sequenzierungstechnologien, Mikrobiologie, Bioinformatik sowie mathematischer Theorie und Modellierung evolutionärer Prozesse ab.
Die Theorie sich schnell verändernder Populationen
Populationsgenetik beschreibt die Dynamik von Mutationen unter dem Einfluss von natürlicher Selektion, Rekombination und den vielfältigen zufälligen Prozessen im Lebenszyklus der Individuen. Traditionell befasst sich die Populationsgenetik mit der Evolution multizellulärer Eukaryonten. Typischerweise wird angenommen, dass die Mehrheit aller beobachteten Mutationen keinerlei Effekt auf den Phänotyp hat und sich nur gelegentlich diejenige Mutation durchsetzt, die Eigenschaften zum Positiven verändert. In Mikroorganismen und insbesondere in RNA-Viren, wozu die Grippe-Viren gehören, sind diese Annahmen nicht gerechtfertigt. Somit wird eine Theorie benötigt, die genau dieser Dynamik schnell adaptierender Populationen Rechnung trägt.
Die Max-Planck-Forschungsgruppe und weitere Wissenschaftlerinnen und Wissenschaftler haben in den letzten Jahren Modelle entwickelt, die der Dynamik mikrobieller Populationen Rechnung tragen und Vorhersagen der genetischen Diversität ermöglichen. Abbildung 2 zeigt schematisch die wesentlichen Züge eines solchen Modells. Individuen in der Population unterscheiden sich durch viele Mutationen, und diese genetische Diversität führt zu phänotypischer Diversität, die wiederum zu Diversität im Replikationserfolg, also der Fitness, führt. Da fitte Individuen im Mittel mehr Nachkommen haben, kommen die Vorfahren der Population typischerweise vom oberen Ende der Fitnessverteilung, während durchschnittliche Individuen auf lange Sicht keine Nachkommen hinterlassen. Der Wettstreit dieser Varianten kann mathematisch beschrieben werden. Diese Theorie liefert explizite Vorhersagen für die Eigenschaften der Stammbäume, die dann mit Sequenzdaten verglichen werden können.
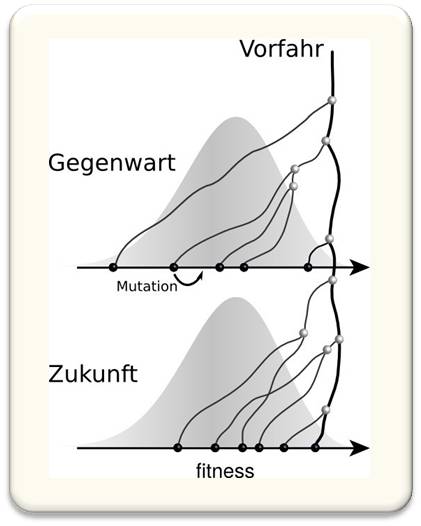 Abb. 2: Schnell evolvierende Populationen sind divers und nur die fittesten Individuen setzen sich durch. Die Vorfahren zukünftiger Populationen kommen vom oberen Rand der Fitnessverteilung, wo seltene Ereignisse die Dynamik dominieren. © Max-Planck-Institut für Entwicklungsbiologie.
Abb. 2: Schnell evolvierende Populationen sind divers und nur die fittesten Individuen setzen sich durch. Die Vorfahren zukünftiger Populationen kommen vom oberen Rand der Fitnessverteilung, wo seltene Ereignisse die Dynamik dominieren. © Max-Planck-Institut für Entwicklungsbiologie.
Die Evolution von Grippe-Viren ist vorhersehbar
Durch das Studium der Populationsmodelle haben wir verstanden, wie tendenziell erfolgreiche, also fitte Viren von weniger erfolgreichen anhand ihrer Genom-Sequenzen unterschieden werden können. Diese Vorhersagen beruhen auf Mustern im Stammbaum der Viren, der aus den Sequenzen rekonstruiert werden kann. Da die erfolgreichen Viren zukünftige Populationen dominieren, erlaubt diese Einsicht Vorhersagen über die Zusammensetzung zukünftiger Populationen.
Für Grippe-Viren sind zuverlässige Vorhersagen besonders relevant, da der Impfstoff gegen Influenza fast jedes Jahr aktualisiert werden muss. Innerhalb weniger Jahre können sich die Viren so stark verändern, dass veraltete Impfstoffe nicht mehr schützen. Die Produktion eines neuen Impfstoffes dauert allerdings mehr als ein halbes Jahr, sodass die Entscheidung über die Impfstoffzusammensetzung lange im Voraus getroffen werden muss. Hier können Vorhersagen, basierend auf einer auf Grippe-Viren bezogenen, neuen Evolutionstheorie, konkret helfen.
Die Forschungsgruppe hat die Vorhersagen ihrer Theorie mit der Evolution von Grippe-Viren zwischen 1995 und 2014 verglichen. Es zeigte sich, dass in fast allen Jahren die Methode eine Virusvariante identifizierte, die nahe an der zukünftigen, tatsächlich aufgetretenen Population lag – in vielen Jahren fiel die Wahl sogar auf die beste Virusvariante. Momentan verfeinern die Wissenschaftlerinnen und Wissenschaftler diese Methode, um der Weltgesundheitsorganisation (WHO) optimale Prognosen für die Impfstoffzusammensetzung zu liefern.
Evolution von HIV im Patienten
Ein weiteres Beispiel schneller Evolution ist die Anpassung von HIV an das individuelle Immunsystem des Wirts. Dieser Prozess passiert jedes Mal wieder, wenn sich ein Mensch mit HIV infiziert. Um diesen Prozess im Detail zu studieren, haben die Max-Planck-Forscher zusammen mit Jan Albert vom Karolinska Institut in Stockholm archivierte Proben von elf Patienten untersucht. Für jeden dieser Patienten standen ihnen fünf bis zwölf Proben zur Verfügung, die die HIV-Infektion von Beginn an über viele Jahre hinweg repräsentieren. Jede Probe enthält Tausende HIV-Genome, die die Wissenschaftlerinnen und Wissenschaftler mit modernen Sequenzierungsmethoden entschlüsseln konnten. Solche Zeitserien von Proben sind im Grunde wie Filme zu betrachten, dank derer die Forscher nachvollziehen können, wie sich die Virus-Population über die Zeit verändert hat.
Abbildung 3 zeigt die Dynamik von Mutationen in einem kleinen Abschnitt des HIV-Genoms innerhalb eines HIV-positiven Menschen. Innerhalb der 350 Basen werden in diesem Beispiel zehn Mutationen beobachtet, die sich durchsetzen. Noch sehr viel mehr Mutationen findet man als seltene Varianten unter 10% in der Population. Anhand solcher Daten konnten die Forscher zum Beispiel zeigen, dass HIV eine Art optimale Sequenz hat und rund ein Drittel aller Veränderungen Reversionen zurück zu dieser optimalen Sequenz darstellen. 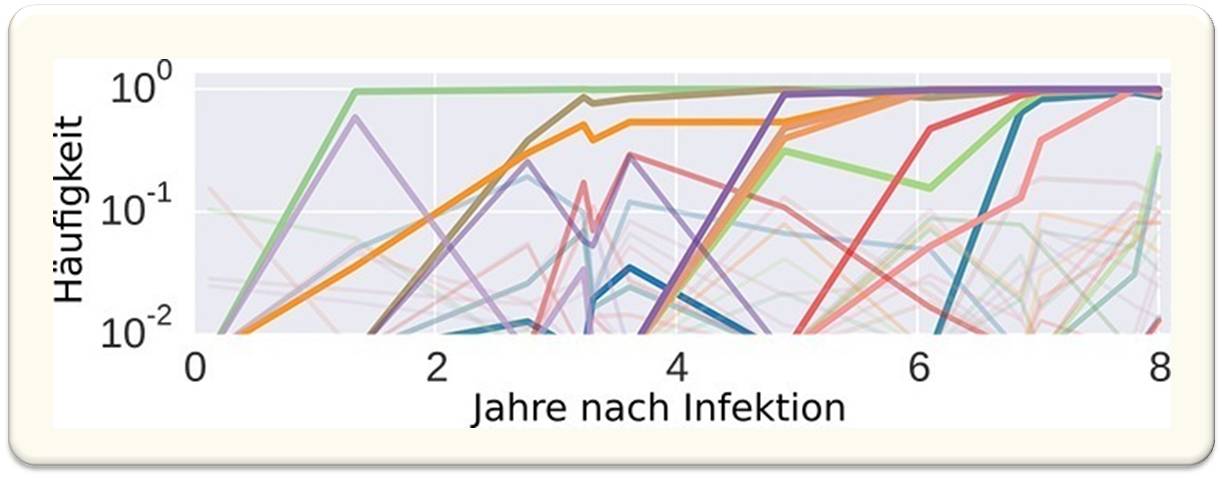
Abb. 3: Die Dynamik von Mutationen im HIV-Protein p17 innerhalb eines HIV-positiven Patienten. Viele Mutationen setzen sich in der Population durch, noch sehr viel mehr erscheinen nur transient. © Max-Planck-Institut für Entwicklungsbiologie, verändert.
Ursprünglich sind diese suboptimalen Varianten vermutlich entstanden, als das Virus dem Immunsystem ausweichen musste. In einem neuen Wirt mit einem anderen Immunsystem können, so wird angenommen, solche Veränderungen dann rückgängig gemacht werden. Diese und andere Resultate zeigen, dass HIV-Populationen problemlos jede mögliche Mutation entdecken können und aus der Fülle an möglichen Mutationen fast deterministisch jene selektieren, die die Virusreplikation insgesamt beschleunigen. Diese Annahme zu prüfen und weitere Methoden zur Vorhersage von Virus-Evolution zu entwickeln, wird die Forscher in Zukunft weiter beschäftigen.
* Der, unter dem Titel "Ist Evolution vorhersehbar? " im Jahrbuch der Max-Planck Gesellschaft 2016 erschienene Artikel (http://www.mpg.de/9826795/MPI_EB_JB_2016) wurde mit freundlicher Zustimmung des Autors und der MPG-Pressestelle ScienceBlog.at zur Verfügung gestellt. Er erscheint hier geringfügig für den Blog adaptiert und ohne die im Original ersichtlichen, nicht frei zugänglichen Literaturstellen (diese können auf Anfrage zugesandt werden).
Weiterführende Links
- Max-Planck-Institut für Entwicklungsbiologie, Tübingen
- Real-time tracking of influenza virus evolution
- neherlab: Blog (englisch) Evolution, population genetics, and infectious diseases
- Infektionen jetten um die Welt, Video 5:56 min (ein mathematisches Modell simuliert, wie Erreger - Beispiel SARS - um den Globus wandern).
- Infektionen auf dem Vormarsch, Video 5:26 min.
Artikel im ScienceBlog
- Peter Schuster; 24.05.2013: Letale Mutagenese — Strategie im Kampf gegen Viren
- Peter Palese; 10.05.2013: Influenza-Viren – Pandemien: sind universell wirksame Impfstoffe in Reichweite?
- Gottfried Schatz; 31.05.2013: Planet der Mikroben — Warum wir Infektionskrankheiten nie endgültig besiegen werden
- Gottfried Schatz; 05.12.2014: Gefahr aus dem Dschungel – Unser Kampf gegen das Ebola-Virus
- Printer-friendly version
- Log in to post comments