Do, 07.07.2017 - 10:23 — Stefan W. Hell

![]() Feinere Details als die halbe Lichtwellenlänge, so war eigentlich seit dem 19. Jahrhundert bekannt, lassen sich im Mikroskop wegen der Lichtbeugung nicht auflösen. Heute steht jedoch fest, dass man mit herkömmlicher Optik fluoreszierende Proben mit einer Detailschärfe weit unterhalb dieser sogenannten Beugungsgrenze abbilden kann. Die Stimulated Emission Depletion-Mikroskopie (STED) und weitere, jüngere fernfeldoptische Verfahren können Auflösungen von besser als 20 Nanometern erreichen und sind prinzipiell sogar in der Lage, molekular auflösen. Der Physiker Stefan Hell (Direktor am Max-Planck-Institut für biophysikalische Chemie/ Göttingen) hat mit der von ihm entwickelten STED-Mikroskopie den minimal-invasiven Zugang zur Nanoskala der Zelle eröffnet. Für die Entwicklung der Fluoreszenz-Nanoskopie wurde 2014 der Nobelpreis für Chemie an Stefan Hell Eric Betzig und William Moerner verliehen.*
Feinere Details als die halbe Lichtwellenlänge, so war eigentlich seit dem 19. Jahrhundert bekannt, lassen sich im Mikroskop wegen der Lichtbeugung nicht auflösen. Heute steht jedoch fest, dass man mit herkömmlicher Optik fluoreszierende Proben mit einer Detailschärfe weit unterhalb dieser sogenannten Beugungsgrenze abbilden kann. Die Stimulated Emission Depletion-Mikroskopie (STED) und weitere, jüngere fernfeldoptische Verfahren können Auflösungen von besser als 20 Nanometern erreichen und sind prinzipiell sogar in der Lage, molekular auflösen. Der Physiker Stefan Hell (Direktor am Max-Planck-Institut für biophysikalische Chemie/ Göttingen) hat mit der von ihm entwickelten STED-Mikroskopie den minimal-invasiven Zugang zur Nanoskala der Zelle eröffnet. Für die Entwicklung der Fluoreszenz-Nanoskopie wurde 2014 der Nobelpreis für Chemie an Stefan Hell Eric Betzig und William Moerner verliehen.*
Sehen ist die für uns wahrscheinlich wichtigste Sinneswahrnehmung. Nicht nur im täglichen Leben "glaubt man nur dem, was man sieht" und "weiß, dass Bilder mehr sagen als 1000 Worte", dies trifft auch auf die modernen Naturwissenschaften zu. Es ist sicherlich kein Zufall, dass deren Beginn mit der Erfindung der Lichtmikroskopie einhergeht. Damit war der Mensch erstmals in der Lage zu sehen, dass jedes lebende System aus Zellen, den Grundeinheiten von Struktur und Funktion, besteht. Jeder von uns hat in der Schule auch sicherlich gelernt, dass die Auflösung des Lichtmikroskops grundsätzlich durch die Wellenlänge des verwendeten Lichts begrenzt ist und bei rund 200 – 350 Nanometern liegt. Will man kleinere Strukturen sehen - beispielsweise Viren -, so benötigt man dazu das Elektronenmikroskop (Abbildung 1).
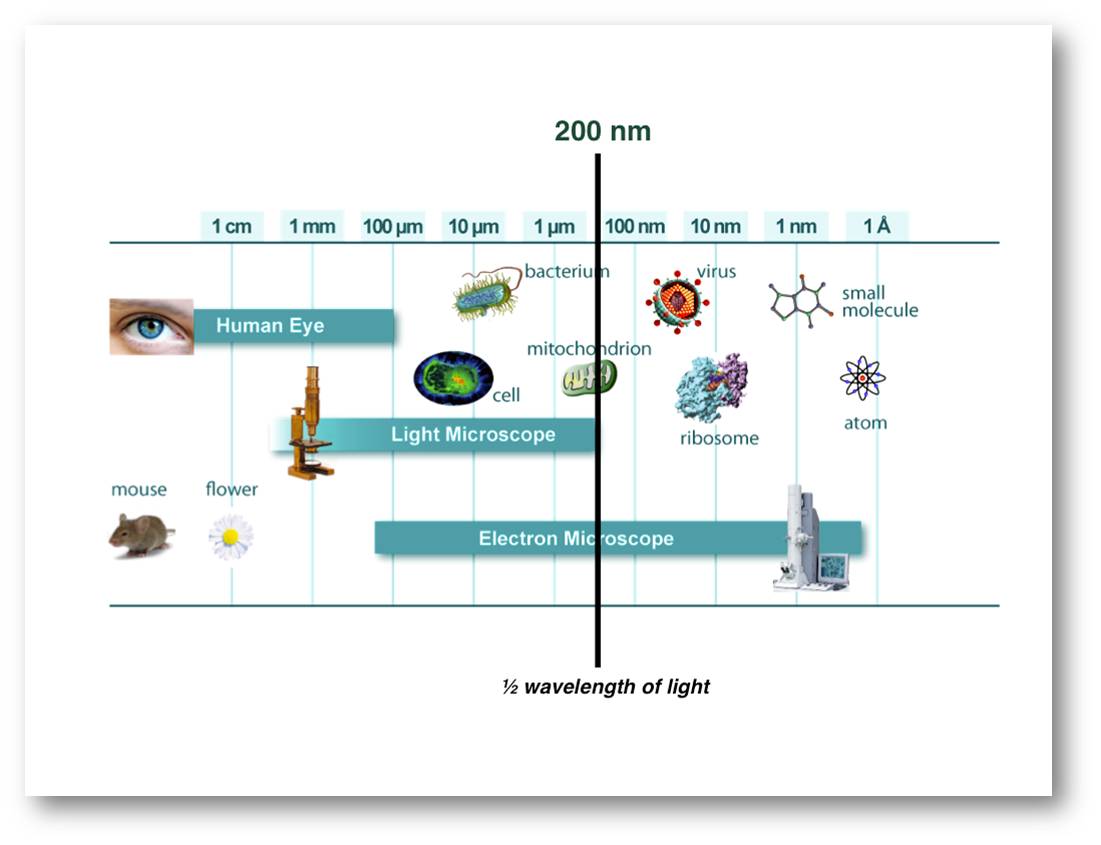 Abbildung 1. Größenskalen und Auflösungsgrenzen für das menschliche Auge, das Lichtmikroskop und das Elektronenmikroskop. Die Auflösungsgrenze des Lichtmikroskops liegt bei rund 200 nm, der halben Wellenlänge des eingestrahlten Lichts (nm: Nanometer = Millionstel Millimeter, µm: Mikrometer = Tausendstel mm).
Abbildung 1. Größenskalen und Auflösungsgrenzen für das menschliche Auge, das Lichtmikroskop und das Elektronenmikroskop. Die Auflösungsgrenze des Lichtmikroskops liegt bei rund 200 nm, der halben Wellenlänge des eingestrahlten Lichts (nm: Nanometer = Millionstel Millimeter, µm: Mikrometer = Tausendstel mm).
Das Elektronenmikroskop wurde in der Mitte des 20. Jahrhunderts erfunden. Es ermöglicht eine um Größenordnungen höhere räumliche Auflösung - in manchen Fällen bis hinab zur Größe eines Atoms - und hat zu zahllosen grundlegenden Entdeckungen geführt.
Wenn wir also mit dem Elektronenmikroskop eine derart hohe Auflösung erzielen…
…warum ist dann die Lichtmikroskopie so wichtig?
Nimmt man erfolgreiche Top-Journale in den Lebenswissenschaften zur Hand und zählt die Untersuchungen, in welchen Mikroskopie angewandt wurde, so findet man, dass im überwiegenden Teil der Fälle die Lichtmikroskopie genutzt wurde, da diese die bei weitem populärste Methode ist. Dafür gibt es zwei wesentliche Gründe:
- Lichtmikroskopie ist die einzige Form der Mikroskopie, mit der man lebende Zellen in allen Raumrichtungen beobachten kann, und sie ist minimal invasiv. Beispielsweise kann man verfolgen, wie Biomoleküle in den Zellen interagieren oder ihre Position verändern - dies ist mit Elektronenmikroskopie nicht möglich.
- Üblicherweise wollen wir wissen, wo sich ein bestimmtes Biomolekül, ein Protein, zu einem bestimmten Zeitpunkt aufhält und ob es mit irgendetwas Anderem interagiert. Weil die vielen Tausende Proteine einer Zelle unter Lichteinfall alle gleich aussehen, muss man das zu untersuchende Protein spezifisch markieren. Dies funktioniert in der Lichtmikroskopie viel einfacher als in der Elektronenmikroskopie: man hängt ein fluoreszierendes Molekül an das Protein und - bei Bestrahlung mit Licht passender Wellenlänge - kann man das Protein anhand seiner Fluoreszenz verfolgen.
Fluoreszierende Moleküle als Marker
existieren u.a. in zwei Zuständen: einem Grundzustand (So) und einem angeregten Zustand (S1) mit höherer Energie. Bei Bestrahlung mit Licht passender Wellenlänge (hier: grünes Licht), nimmt das Molekül ein (grünes) Photon auf und geht vom Grundzustand in den angeregten Zustand S1 über. Etwas von dieser Energie geht durch Schwingungen der Atome verloren - das Molekül fällt auf ein tieferes S1-Niveau und kehrt von dort unter Aussenden eines Photons innerhalb von Nanosekunden in seinen Grundzustand So zurück. Infolge des Energieverlusts ist die Wellenlänge des emittierten Photons in den längerwelligen Bereich verschoben (Abbildung 2).
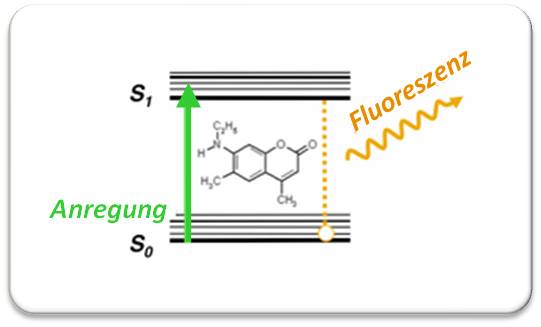 Abbildung 2. Energieschema eines Fluoreszenzmarkers (hier ein Cumarinfarbstoff). Der Marker wird durch Licht (grün) in einen Zustand S1 angeregt, verliert etwas an Energie und relaxiert (geht über) von einem niedrigeren S1-Niveau unter Emission von längerwelligem Fluoreszenzlicht (orange) in den Grundzustand. Aufgrund der unterschiedlichen Wellenlängen kann das Fluoreszenzlicht separiert vom Anregungslicht betrachtet werden. Dies macht Fluoreszenzmessungen enorm sensitiv, man kann auch noch ein einzelnes, mit einem Fluoreszenzmarker versehenes Biomolekül in der Zelle detektieren.
Abbildung 2. Energieschema eines Fluoreszenzmarkers (hier ein Cumarinfarbstoff). Der Marker wird durch Licht (grün) in einen Zustand S1 angeregt, verliert etwas an Energie und relaxiert (geht über) von einem niedrigeren S1-Niveau unter Emission von längerwelligem Fluoreszenzlicht (orange) in den Grundzustand. Aufgrund der unterschiedlichen Wellenlängen kann das Fluoreszenzlicht separiert vom Anregungslicht betrachtet werden. Dies macht Fluoreszenzmessungen enorm sensitiv, man kann auch noch ein einzelnes, mit einem Fluoreszenzmarker versehenes Biomolekül in der Zelle detektieren.
Sensitivität darf aber nicht mit Auflösung verwechselt werden!
Auflösung ist etwas Anderes, es bedeutet, dass man individuelle Strukturen voneinander getrennt wahrnimmt. Dies ist in der Lichtmikroskopie nicht mehr der Fall, wenn der Abstand der einzelnen Strukturen/Moleküle weniger als 200 nm beträgt. Dann erscheinen diese als ein einziger verschwimmender Lichtfleck.
Ein Fluoreszenzmikroskop mit einer wesentlich höheren räumlichen Auflösung, die bis in den Bereich der Biomoleküle reicht, sollte daher einen ungeheuren Einfluss auf die Naturwissenschaften und darüber hinaus haben.
Wodurch wird die Auflösung in der Lichtmikroskopie begrenzt?
Das wichtigste Element in der Lichtmikroskopie ist die Linse des Objektivs. Diese hat die Aufgabe, das Licht in einem Punkt zu bündeln. Da Licht sich aber in Form einer Welle fortpflanzt, kann die Linse dieses nicht an einem einzelnen geometrischen Punkt scharf bündeln - das Licht wird gebeugt und es entsteht ein Lichtfleck, der mindestens 200 nm breit und (entlang der optischen Achse) 500 nm lang ist. Dies hat zur Folge, dass alle Strukturen, die sich innerhalb dieses Flecks befinden, gleichzeitig bestrahlt werden und gleichzeitig Fluoreszenzlicht emittieren. Kein wie auch immer gearteter Detektor kann aufgrund der Beugungsgrenze diese überlappenden Signale voneinander trennen (Abbildung 3).
Dass Lichtbeugung eine fundamentale Grenze für die optische Auflösung setzt, wurde von Ernst Abbe (1840 -1905) erkannt. Abbe hat die nach ihm benannte Formel aufgestellt, die in allen Lehrbüchern der Physik, Optik und Lebenswissenschaften zu finden ist. Diese besagt: um zwei ähnliche Strukturen getrennt beobachten zu können, müssen diese voneinander weiter getrennt liegen als die Wellenlänge des Lichts λ geteilt durch die zweifache numerische Apertur (diese beinhaltet den halben Öffnungswinkel λ und den Brechungsindex n) des Objektivs (Abbildung 3).
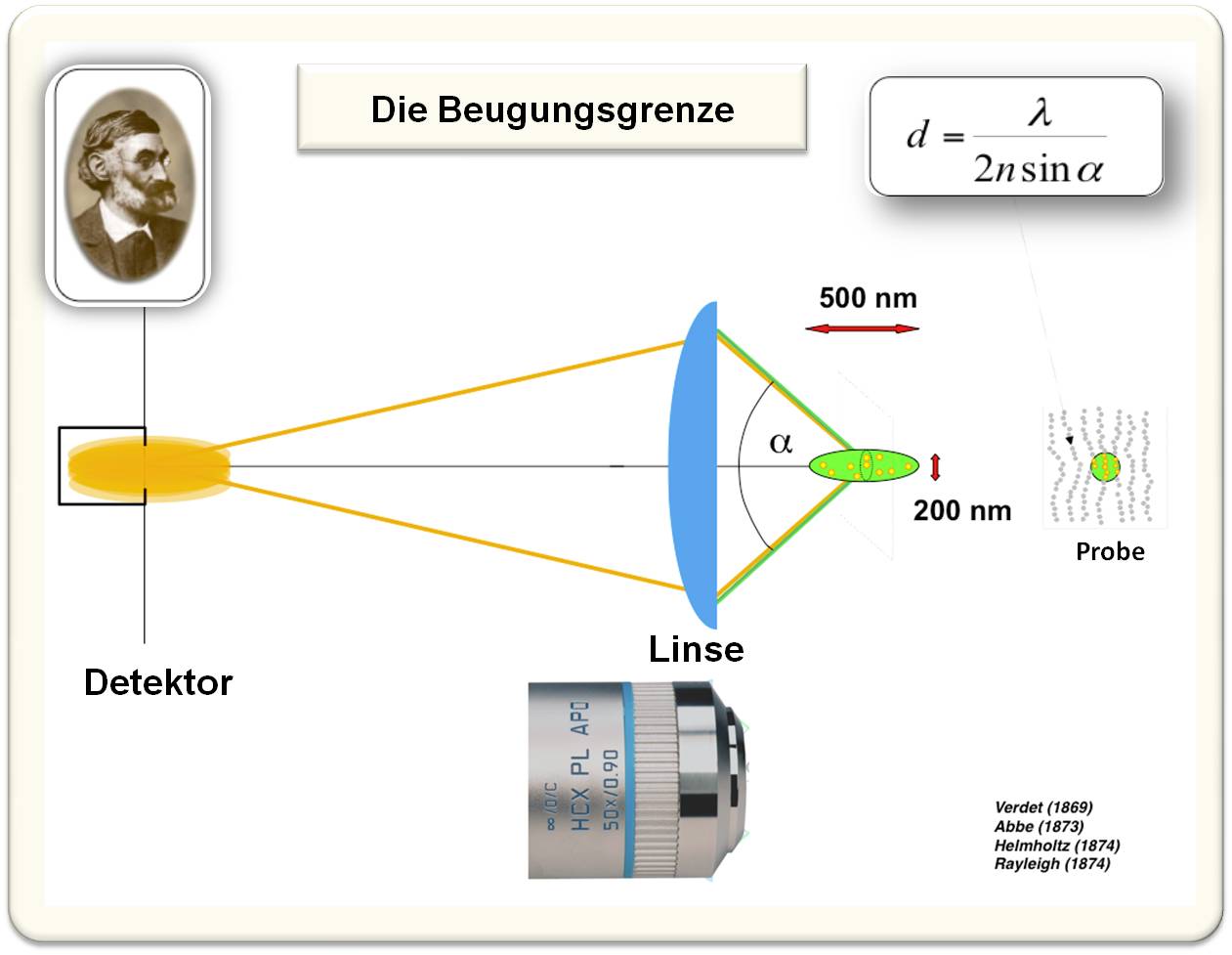 Abbildung 3. Zur Beugungsgrenze: Die Linse eines Objektivs kann Licht nicht in einem Punkt fokussieren. Um zwei ähnliche Strukturen getrennt wahrnehmen zu können, müssen diese mindestens um die Hälfte der eingestrahlten Wellenlänge λ voneinander entfernt sein. Ernst Abbe (links oben) hat dies in der nach ihm benannten Formel für die Beugungsgrenze (d, rechts oben) festgelegt. (n: Brechungsindex).
Abbildung 3. Zur Beugungsgrenze: Die Linse eines Objektivs kann Licht nicht in einem Punkt fokussieren. Um zwei ähnliche Strukturen getrennt wahrnehmen zu können, müssen diese mindestens um die Hälfte der eingestrahlten Wellenlänge λ voneinander entfernt sein. Ernst Abbe (links oben) hat dies in der nach ihm benannten Formel für die Beugungsgrenze (d, rechts oben) festgelegt. (n: Brechungsindex).
An die Gültigkeit dieser Formel haben auch alle Physiker und Lebenswissenschafter des 20. Jahrhunderts geglaubt. Vielleicht, so dachte man, ließe sich diese Auflösung noch um einen Faktor 2 verbessern, damit wäre dann aber Schluss.
Als Student in Heidelberg war für Stefan Hell in den späten 1980er Jahren die Grenze der optischen Auflösung noch ein Faktum.
Kann die Beugungsgrenze durchbrochen werden?
Viele, wenn nicht die meisten Entdeckungen sind mit den Lebensumständen ihrer Entdecker verknüpft. Hell war mit seinen Eltern von Osteuropa - dem Banat in Rumänien - nach Westeuropa - Ludwigshafen - gezogen und hatte in Heidelberg Physik studiert. Seine Doktorarbeit hatte er bei einem Physiker begonnen, der zusammen mit einem Kollegen am Institut eine Start-up Firma gründete, welche Beziehungen zu IBM und Siemens hatte. Ziel war die Entwicklung von lichtmikroskopischen Methoden - genauer gesagt von konfokaler Mikroskopie - zur Prüfung von Computer-Chips.
Nach einem Jahr Arbeit an dem Thema wuchs Hells Frustration. Für jemanden, der an Grundlagenforschung interessiert war, gab es in der Lichtmikroskopie nichts Neues mehr, nur Linsen und Fokussieren von Licht - es war die Physik des 19. Jahrhunderts. Das einzig Interessante und Wichtige – so dachte er – würde wohl sein, die Beugungsgrenze zu durchbrechen. Er war überzeugt, dass es dafür einen Weg geben müsse. Die Beugungsgrenze war 1873 definiert worden, seitdem war in mehr als 100 Jahren so viel Neues in der Physik dazu gekommen - Quantenmechanik, Quantenoptik, Moleküle und ihre Zustände - es musste zumindest ein physikalisches Phänomen existieren, mit dem sich die Beugungsgrenze austricksen ließe.
In der Hoffnung derartige Phänomene zu entdecken, wälzte Hell Lehrbücher und versuchte Kollegen zu überzeugen, dass es wert wäre dieses Problem anzugehen, möglicherweise von den Eigenschaften der Fluorophore aus. Es bestand in Heidelberg aber kein Interesse, und für Hell bestand die Aussicht ohne Job da zu stehen.
Ein finnischer Kollege schlug ihm schließlich vor zu einem ihm bekannten Professor nach Finnland zu gehen. Dieser würde ihm den Freiraum bieten an seinen Ideen zur Auflösungsgrenze zu arbeiten; falls dies glücken sollte, würden ihn die Deutschen schon bitten zurückzukommen. Hell landete dann mit einem Stipendium der Finnischen Akademie in Turku als ein unabhängiger Postdoc. Als er eines Morgens im Jahr 1993 über Quantenphänomene des Lichts las, blieb er an einer Seite hängen, die von stimulierter Emission handelte. Mit diesem Phänomen, das jeder Physiker in seinem ersten Studienjahr kennenlernt, sollte es möglich sein die Auflösungsgrenze zu umgehen - zumindest was Fluoreszenzmessungen betrifft.
Das Problem der optischen Auflösung war ja, dass eine Linse keinen Lichtfleck produzieren kann, der schärfer als 200 nm ist und alle Moleküle, die in diesem Fleck durch Licht angeregt werden, dann zusammen Fluoreszenzlicht emittieren. Sollte es allerdings gelingen einen Teil der Moleküle in einen Zustand zu bringen, in dem sie kein Licht emittieren können, dann würde die Fluoreszenz von einem engeren räumlichen Bereich als dem Anregungsspot kommen und damit eine höhere Auflösung erbringen.
Die Schlüsselidee war also nicht an der Linse herum zu probieren, sondern mit dem Energiezustand der Moleküle zu spielen.
Das Konzept: ein physikalischer Trick mittels "Stimulierter Emissionsauslöschung (STED)"
In einem Fluorophor gibt es einen Grundzustand und einen angeregten Zustand (s. Abbildung 2). Der letztere ist ein heller Zustand, das Molekül emittiert ein Photon. Im Grundzustand kann das Molekül kein Photon erzeugen, es ist ein dunkler Zustand. Licht kann nun nicht nur ein Molekül anregen, sondern auch ein angeregtes Molekül schlagartig abregen und zwar durch 'stimulierte Emission', einen Prozess, der bereits von Einstein vorhergesagt worden war (Abbildung 4a).
Werden (mit z.B grünem Licht) die Fluorophore angeregt, so emittieren diese normalerweise längerwelliges – hier orange dargestelltes – Fluoreszenzlicht, aus dem grünen Lichtfleck, dessen Auflösung durch die Lichtbeugung limitiert ist. Wird nun aber gleichzeitig längerwelliges (rot dargestelltes) Licht eingestrahlt, so werden die Moleküle in den dunklen Zustand überführt. Ab einem Schwellwert der Intensität (Is) des stimulierenden roten Lichtes wird die Fluoreszenz praktisch vollkommen abgeschaltet.
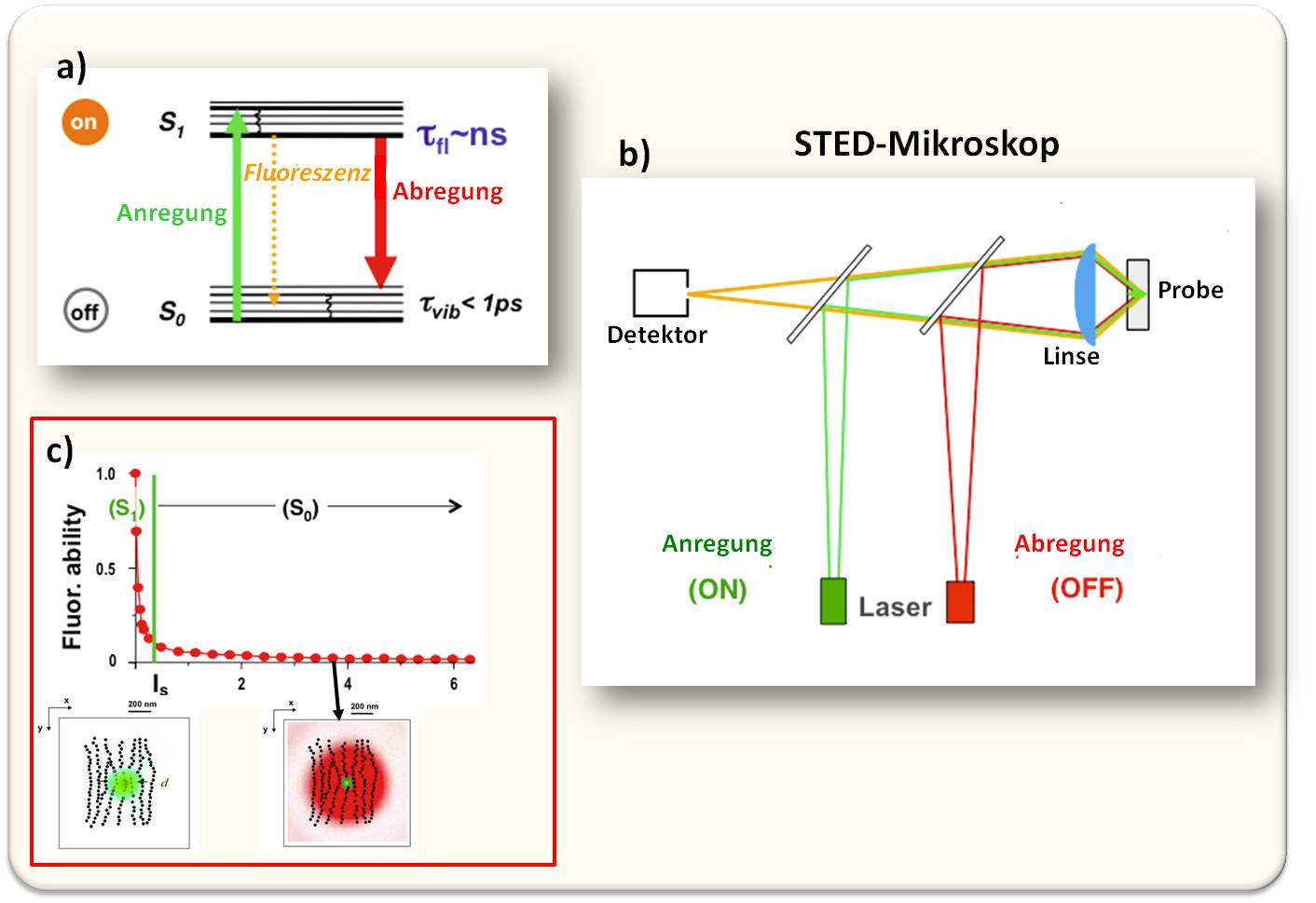 Abbildung 4. Prinzip des STED-Mikroskops (STimulated Emission Depletion - Stimulierte Emissionsauslöschung). a) Anregung der Fluoreszenzemission durch einen grünen Lichtpuls, und Abregung durch einen roten Lichtpuls. (Die Fluoreszenzemission erfolgt im Bereich von Nanosekunden, die Molekülschwingungen in Pikosekunden) b) Schema des STED-Mikroskops: in der Brennebene des Objektivs überlappen synchron Anregungslichtpuls und doughnut-förmiger Abregungslichtpuls. c) Die Wahrscheinlichkeit für ein Molekül, sich im angeregten hellen Zustand (S1) zu befinden. Oberhalb eines Schwellwertes seiner Intensität Is schaltet der doughnut-förmige Abregungslichtpuls die Fluoreszenzemission de facto ab - nur in seinem Zentrum bleibt der S1-Zustand erlaubt, Moleküle können dort fluoreszieren, und dies führt damit zu Bildern weit unterhalb der Beugungsgrenze. (Hell & Wichmann, 1994 Opt. Letters)
Abbildung 4. Prinzip des STED-Mikroskops (STimulated Emission Depletion - Stimulierte Emissionsauslöschung). a) Anregung der Fluoreszenzemission durch einen grünen Lichtpuls, und Abregung durch einen roten Lichtpuls. (Die Fluoreszenzemission erfolgt im Bereich von Nanosekunden, die Molekülschwingungen in Pikosekunden) b) Schema des STED-Mikroskops: in der Brennebene des Objektivs überlappen synchron Anregungslichtpuls und doughnut-förmiger Abregungslichtpuls. c) Die Wahrscheinlichkeit für ein Molekül, sich im angeregten hellen Zustand (S1) zu befinden. Oberhalb eines Schwellwertes seiner Intensität Is schaltet der doughnut-förmige Abregungslichtpuls die Fluoreszenzemission de facto ab - nur in seinem Zentrum bleibt der S1-Zustand erlaubt, Moleküle können dort fluoreszieren, und dies führt damit zu Bildern weit unterhalb der Beugungsgrenze. (Hell & Wichmann, 1994 Opt. Letters)
Nun kann man sich bereits vorstellen, wie ein mit stimulierter Emissionsauslöschung arbeitendes Mikroskop funktioniert: Es wird nur ein Teil der Fluoreszenzemission abgeschaltet. Am besten gelingt dies, wenn der rote Lichtstrahl einen doughnut-förmigen Ring im Außenbereich des grünen Anregungsspots bildet - die Emissions-Signale kommen dann nur mehr von den Molekülen im Zentrum. Mit diesem verkleinerten Bereich, für den Moleküle fluoreszieren können, kann man nun über das Probenfeld rastern lassen und das Fluoreszenzlicht mit einer wesentlich höheren Auflösung registrieren als ein normales durch die Lichtbeugung limitiertes Mikroskop (Abbildung 4b, c).
Dieses Konzept hatte Stefan Hell bereits 1994 veröffentlicht - nicht gerade in einem Journal, wie man es von der großen Bedeutung dieser Idee in der Folge erwarten würde - und dann in den USA und in Europa publik zu machen versucht. Er hoffte einen Job, ein Labor angeboten zu bekommen. Man hielt aber die Auflösung im Nanoskalenbereich nicht für möglich. Erst 1997 boten ihm die damaligen Direktoren am Max-Planck-Institut für biophysikalische Chemie in Göttingen (insbesondere auch Tom Jovin, Anm. Red.) die Möglichkeit, als Leiter einer selbstständigen Nachwuchsgruppe dieses Konzept weiter zu erkunden, seine Richtigkeit zu bestätigen.
Die STED-Mikroskopie funktioniert
Es stellte sich heraus, dass das Konzept hervorragend funktionierte. In der Folge erzielten Hell und seine Kollegen Aufnahmen, deren Auflösung weit jenseits der Beugungsgrenze lag. Dies sollen einige Beispiele in Abbildung 5 verdeutlichen.
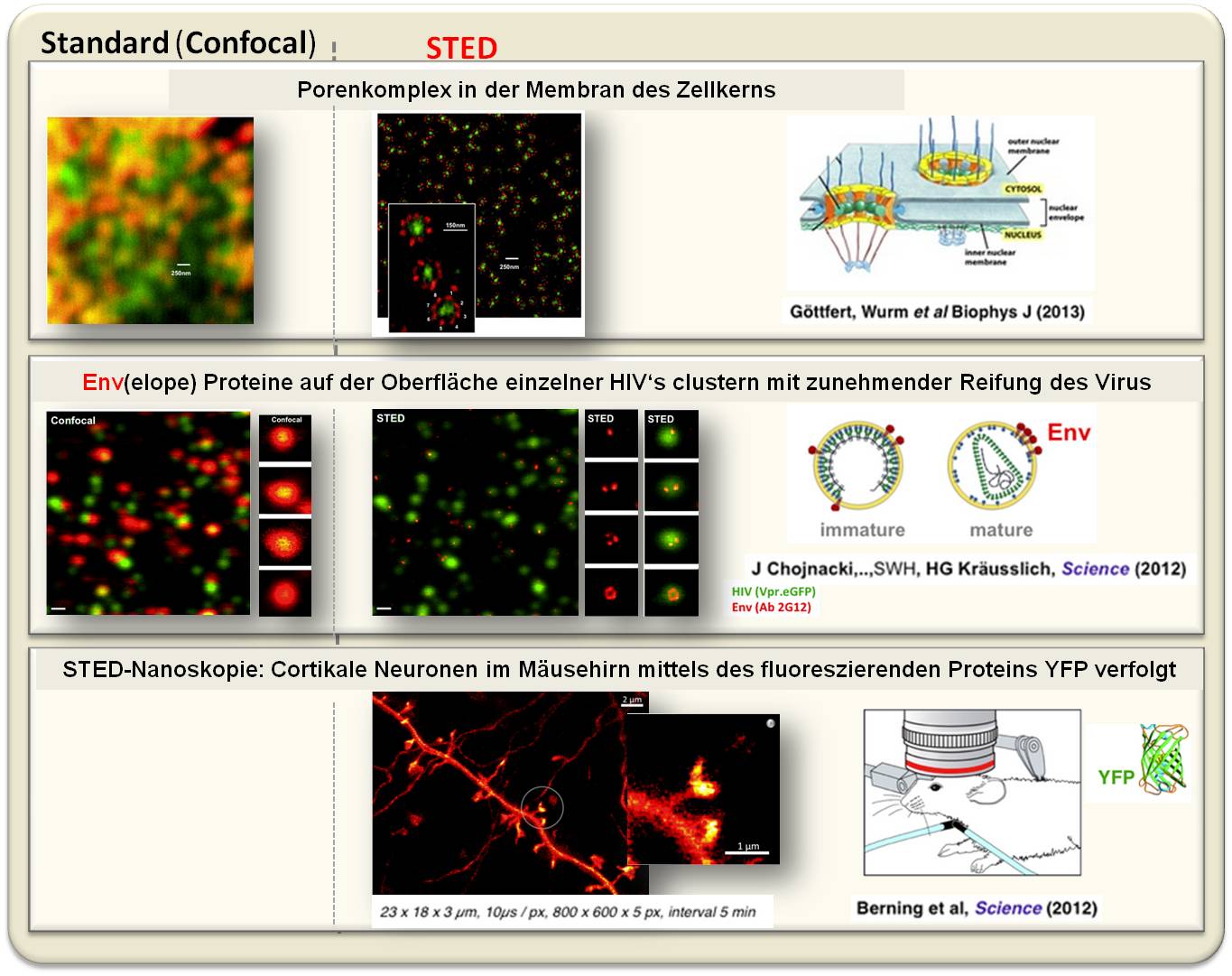 Abbildung 5. Beispiele für die mit dem STED-Mikroskop erhaltene Auflösung. Oben: Architektur der Kernporen in der Membran eines intakten Zellkerns im Vergleich mit konventioneller konfokaler Mikroskopie. Insert: der Ring mit achtfach-symmetrischer Anordnung eines Proteins hat einen Durchmesser von rund 160 nm, ca. 20 nm Auflösung ermöglichen die getrennte Darstellung der Untereinheiten mit STED-Nanoskopie. Mitte: Das Hüllprotein Env (rot) sammelt sich mit zunehmender Reifung des HIV-Virus (grün) auf dessen Oberfläche an einer Stelle. (Reifes Partikel: oben). Unten: Dendriten in der visuellen Hirnrinde einer lebenden Maus mit den synaptischen Enden. Mittels Zeitrafferaufnahmen wurde deren Plastizität gezeigt.
Abbildung 5. Beispiele für die mit dem STED-Mikroskop erhaltene Auflösung. Oben: Architektur der Kernporen in der Membran eines intakten Zellkerns im Vergleich mit konventioneller konfokaler Mikroskopie. Insert: der Ring mit achtfach-symmetrischer Anordnung eines Proteins hat einen Durchmesser von rund 160 nm, ca. 20 nm Auflösung ermöglichen die getrennte Darstellung der Untereinheiten mit STED-Nanoskopie. Mitte: Das Hüllprotein Env (rot) sammelt sich mit zunehmender Reifung des HIV-Virus (grün) auf dessen Oberfläche an einer Stelle. (Reifes Partikel: oben). Unten: Dendriten in der visuellen Hirnrinde einer lebenden Maus mit den synaptischen Enden. Mittels Zeitrafferaufnahmen wurde deren Plastizität gezeigt.
Beispiel aus der Zellbiologie:
Das STED-Bild des Kernporenkomplexes in der Membran des Zellkerns zeigt eine rund zehnmal höhere Auflösung als es mit konventioneller Mikroskopie (links) möglich ist. Die Architektur der Pore mit einer achtfachen Symmetrie der Protein-Untereinheiten (jeweils Durchmesser 20 - 40 nm) wird deutlich.
Beispiel aus der Virologie:
Viren sind für die konventionelle Lichtmikroskopie zu klein (Durchmesser 30 -150 nm). Will man dann noch die Proteine auf der Virushülle sehen, so ist das auch mit der Elektronenmikroskopie sehr schwierig. Beispielsweise sitzen 15 - 40 Kopien des Env(elope) Proteins auf der Oberfläche eines HIV-Virus. Mit Hilfe der STED-Mikroskopie konnten die Forscher zeigen, dass diese Proteine bei Reifung des Virus sich zu einem einzigen Env-Cluster zusammenfinden - offensichtlich eine Voraussetzung für die Infektiösität.
Beispiel aus der Neurophysiologie:
Ein Blick in das Hirngewebe einer lebenden, Gen-manipulierten Maus, deren Neuronen das fluoreszierendes Protein YFP ("yellow fluorescent protein") exprimieren. In 20 µm Tiefe, in der oberen Schichte der visuellen Hirnrinde sieht man einen Teil eines Dendriten mit den sogenannten" Dornenfortsätzen" - den becherförmigen signal-empfangenden Teilen der Synapsen. Zeitrafferaufnahmen in 5-Minuten-Intervallen zeigen, dass sich diese Dornfortsätze offensichtlich verändern, leicht bewegen, dass eine gewisse Plastizität im Gehirn besteht. Diese Ergebnisse geben zum Optimismus Anlass, dass man mittels STED auch die Vorgänge der Signalübertragung direkt an den Synapsen verfolgen wird können.
Das STED-Mikroskop
Nach anfänglichem Herumprobieren und Optimieren der Parameter, mit denen die Beugungsgrenze umgangen wird, ist das STED-Mikroskop nun am Markt erhältlich und wird von drei Firmen angeboten (Abbildung 6). Die Intensität des STED-Strahls kann so eingestellt werden, dass die Ausdehnung des Bereichs, in dem die Moleküle fluoreszieren können, beliebig verringert werden kann. Mit Lichtstrahlen für einen so verengten fluoreszenzfähigen Bereich wird die Probe abgerastert und somit ein Bild erstellt.
 Abbildung 6. Das STED-Mikroskop im Laboraufbau und kommerziell angeboten (u.a. von Abberior Instruments, einer Firma, die Postdocs aus Hells Gruppe in Göttingen gegründet haben).
Abbildung 6. Das STED-Mikroskop im Laboraufbau und kommerziell angeboten (u.a. von Abberior Instruments, einer Firma, die Postdocs aus Hells Gruppe in Göttingen gegründet haben).
Die STED-Mikroskopie lässt sich mit dynamischen Methoden wie der Fluoreszenz-Korrelations-Spektroskopie und Techniken der schnellen Lichtstrahl-Rasterung kombinieren. Auch die Einzelmolekül-Methoden PALM/STORM basieren (wie das STED-Mikroskop) auf dem An- und Ausschalten der Fluoreszenzfähigkeit von Molekülen. Der wesentliche Unterschied in diesem Konzept besteht darin, dass dort nur wenige Moleküle angeregt werden, die jeweils weiter entfernt sind als die Beugungsgrenze von 200 nm und daher getrennt wahrgenommen werden können.
Für die Entwicklung der ultra-hochauflösenden Fluoreszenz-Mikroskopie wurde 2014 der Nobelpreis für Chemie verliehen. Gemeinsam mit Stefan Hell erhielten ihn Eric Betzig und William Moerner.
Wo liegt nun die Grenze der Auflösung?
Um Strukturen voneinander getrennt wahrnehmen zu können, hat man im 20. Jahrhundert versucht Licht so scharf wie möglich zu fokussieren. Auch die besten Linsen waren natürlich durch die Lichtbeugung begrenzt.
Heute wird die erzielte Auflösung durch das An- und Abschalten der fluoreszierenden Moleküle bestimmt, durch Schalten zwischen zwei Zuständen des Moleküls. Der Übergang zwischen zwei Zuständen gilt nicht unbedingt nur für fluoreszierende Moleküle. Es können beispielsweise auch Änderungen der Konformation (z.B. cis-trans-Übergänge, die dann auch fluoreszierend/dunkel sind), oder, denkbarerweise, der Absorption, der Streuung oder auch des Spinzustands als Zustandspaar herangezogen werden (Abbildung 7).
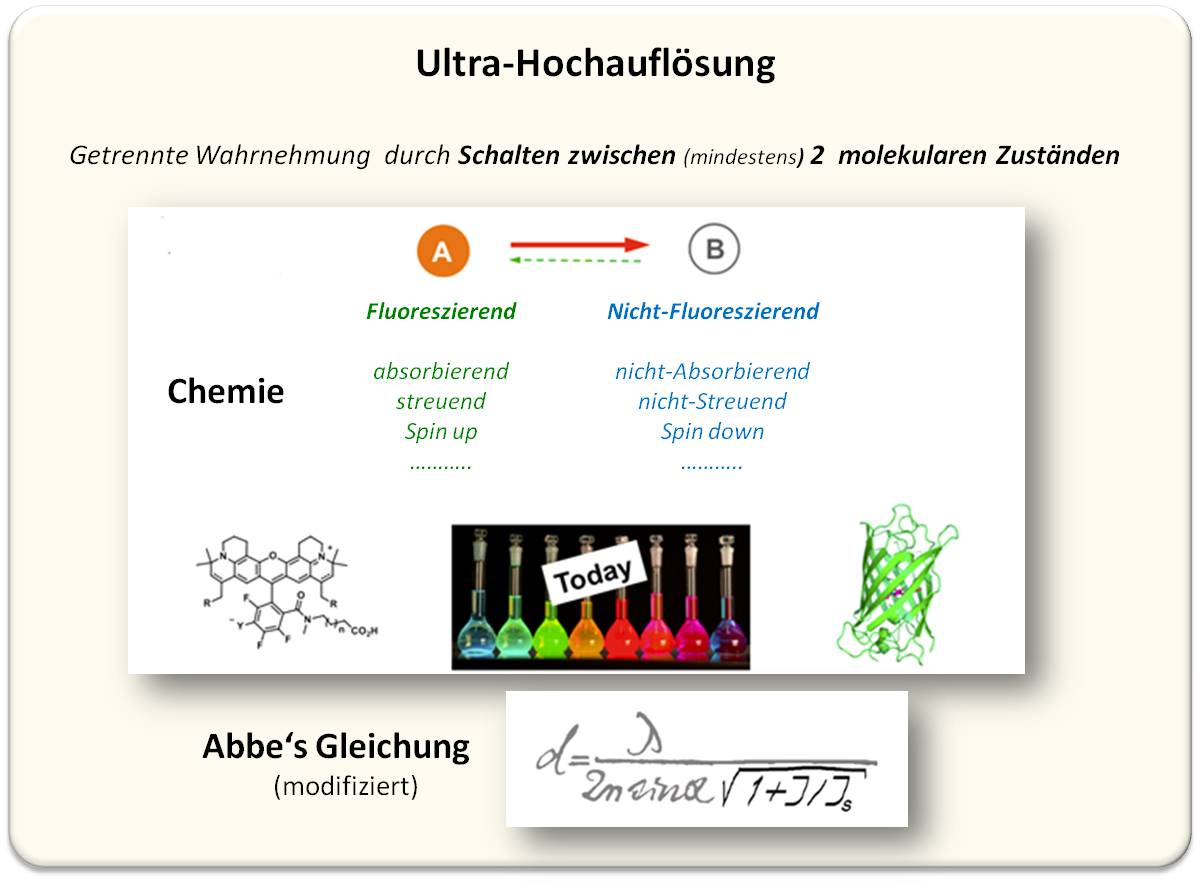 Abbildung 7. Ultra-Hochauflösung wird nicht mehr durch die Qualität der Linse sondern durch die Chemie des Moleküls bestimmt. Eine adaptierte Form von Abbes Formel zur Beugungsgrenze wurde aus dem STED-Konzept hergeleitet (I und Is: siehe Abbildung 4).
Abbildung 7. Ultra-Hochauflösung wird nicht mehr durch die Qualität der Linse sondern durch die Chemie des Moleküls bestimmt. Eine adaptierte Form von Abbes Formel zur Beugungsgrenze wurde aus dem STED-Konzept hergeleitet (I und Is: siehe Abbildung 4).
Heute hängt die Auflösung nicht mehr von der Qualität der Linse sondern von der Chemie des Moleküls ab. Dabei kann prinzipiell eine Auflösung bis in den Größenbereich des Moleküls selbst erzielt werden. Dies geht aus der adaptierten Abbe-Formel hervor, die aus STED-Studien hergeleitet wurde (Die Auflösung wird sehr hoch, wenn der Quotient aus Intensität des STED-Lichtstrahls und Schwellwert-Intensität sehr groß wird; Abbildung 7).
Der Weg zu Mikroskopen, die eine Auflösung bis hinab zur Molekülgröße - und damit ungeahnte Einblicke in die Mechanismen zellulärer Vorgänge - ermöglichen, ist klar. Es ist nur eine Frage der technischen Umsetzung.
*Der vorliegende Artikel ist eine verkürzte, deutsche Fassung des Vortrags "Unlimited sharp: light microscopy in the 21st century", den Stefan Hell am 9. November 2016 anlässlich der Verleihung des Wilhelm-Exner Preises in Wien gehalten hat. Video 45:50 min: https://slideslive.com/38899163/unlimited-sharp-light-microscopy-in-the-...
Weiterführende Links
- Stefan W. Hell: http://www.mpibpc.mpg.de/de/hell
- Stefan W. Hell: Nobel Lecture at Uppsala University (2014). Nanoscopy with focused light. Video 29:06 min.
- The Royal Swedish Academy of Sciences: Information zum Nobelpreis in Chemie 2014: How the optical microscope became a nanoscope.
- Planet Wissen - Mit dem Mikroskop zum Nobelpreis. Video 58:20 min. (Standard-YouTube-Lizenz)
- Stefan Hell (Chemie-Nobelpreis 2014): STED - Lichtblicke in die Nanowelt, Video 7:23 min. (Standard-YouTube-Lizenz)
- Stefan Hell: Neues Gesetz zur Auflösung in der Lichtmikroskopie ermöglicht Bilder in bisher unbekannter Schärfe. Jahrbuch 2005 der Max-Planck-Gesellschaft.
Artikel in ScienceBlog.at:
Redaktion 04.09.2015: Superauflösende Mikroskopie zeigt Aufbau und Dynamik der Bausteine in lebenden Zellen
- Printer-friendly version
- Log in to post comments