Do, 27.07.2017 - 08:35 — Francis S. Collins

![]() Cystische Fibrose (CF) ist in unserer Bevölkerung die häufigste angeborene Stoffwechselerkrankung. Auf Grund einer Genmutation kommt es zum Funktionsverlust des Kanalproteins CFTR, welches in den Zellmembranen von Lunge und anderen Teilen des Körpers das Salz- und Wassergleichgewicht reguliert. In der Folge sammelt sich bereits im frühen Alter dicker, klebriger Schleim an und führt zu schwersten Krankheitserscheinungen und vorzeitigem Tod. Francis Collins, Direktor der US National Institutes of Health (NIH) und ehem. Leiter des "Human Genome Project", hat 1989 das CF verursachende Gen identifiziert. Er beschreibt hier, wie akribische Forschung die Funktionsweise des Proteins CFTR aufklärte und es zur Zielstruktur für das Design von Wirkstoffen machte, die als Kombinationstherapie eben aufsehenerregende klinische Erfolge erzielten.*
Cystische Fibrose (CF) ist in unserer Bevölkerung die häufigste angeborene Stoffwechselerkrankung. Auf Grund einer Genmutation kommt es zum Funktionsverlust des Kanalproteins CFTR, welches in den Zellmembranen von Lunge und anderen Teilen des Körpers das Salz- und Wassergleichgewicht reguliert. In der Folge sammelt sich bereits im frühen Alter dicker, klebriger Schleim an und führt zu schwersten Krankheitserscheinungen und vorzeitigem Tod. Francis Collins, Direktor der US National Institutes of Health (NIH) und ehem. Leiter des "Human Genome Project", hat 1989 das CF verursachende Gen identifiziert. Er beschreibt hier, wie akribische Forschung die Funktionsweise des Proteins CFTR aufklärte und es zur Zielstruktur für das Design von Wirkstoffen machte, die als Kombinationstherapie eben aufsehenerregende klinische Erfolge erzielten.*
Als NIH-Direktor höre ich häufig Erzählungen, wie Menschen mit schweren Erkrankungen - von Arthritis bis hin zu Zika-Infektionen - davon profitieren, dass NIH-Investitionen in die Grundlagenforschung fließen. Heute möchte ich eines der Beispiele bringen, das ich für besonders aufregend halte: es ist die Nachricht, dass eine Kombination von drei, für Zielmoleküle designte Arzneimittel es möglich machen könnte den Großteil aller an cystischer Fibrose (Mukoviszidose) erkrankten Patienten zu therapieren; es ist dies die in unserer Bevölkerung häufigste genetische Erkrankung. Abbildung 1.
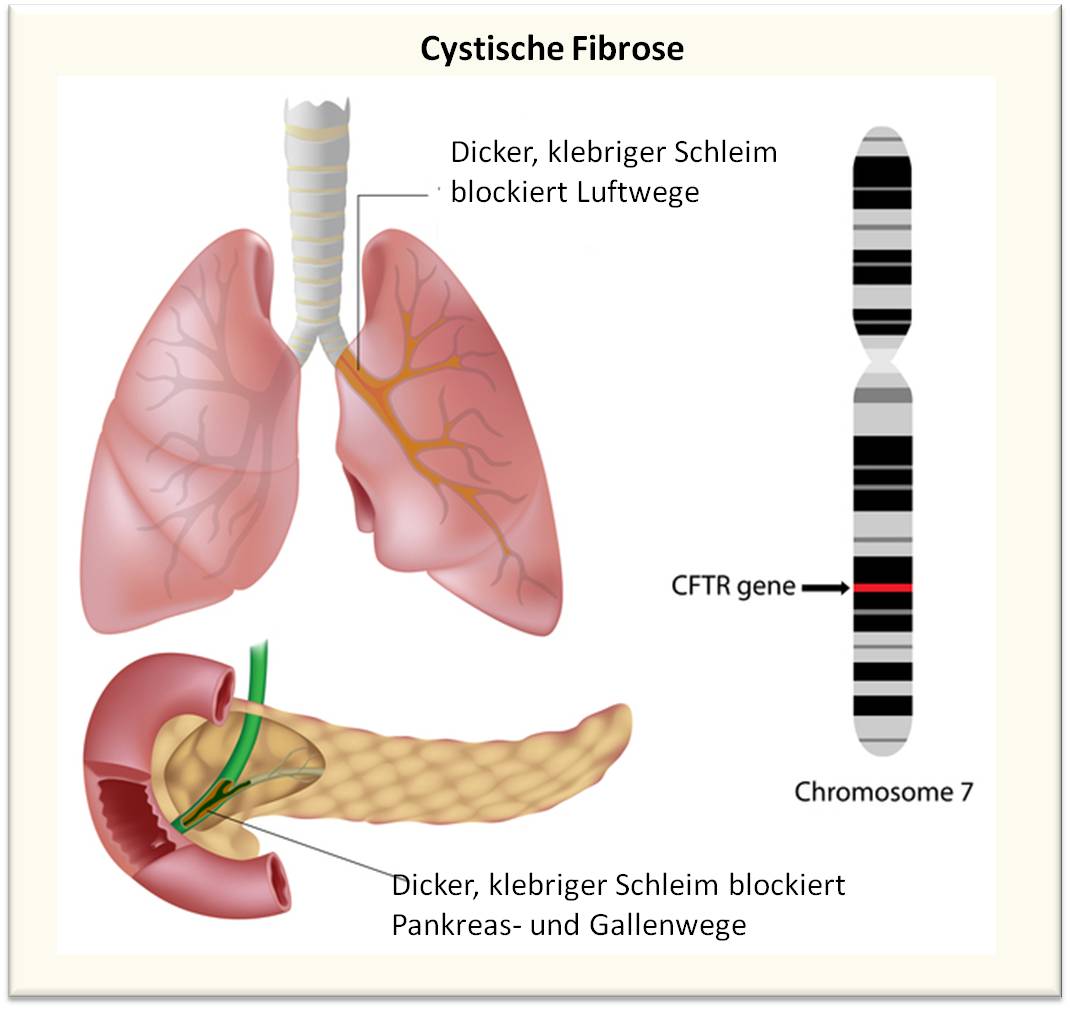
Abbildung 1. Cystische Fibrose ist eine Erbkrankheit der sekretorischen Drüsen verursacht durch Mutationen am CTRF-Gen. Hauptsächlich sind Lunge und Pankreas aber auch Leber, Darm und Geschlechtsorgane betroffen. Dünnflüssiger Mukus, der sezerniert wird, um einige Organe und Körperhöhlen feucht zu halten und zu schützen, wird zu klebrigem Schleim eingedickt, der Luftwege und Sekretionsgänge blockiert. (Bild und Beschriftung von der Redaktion eingefügt; Quelle: https://www.nhlbi.nih.gov/health/health-topics/topics/cf)
Von der Entdeckung der kausalen Ursache zu molekular gezieltem Design von Wirkstoffen
Vorweg etwas zur Geschichte der cystischen Fibrose.
Die erste genetische Mutation, die cystische Fibrose (CF) hervorruft, wurde vor fast 30 Jahren entdeckt - es war eine Zusammenarbeit meines eigenen Forschungslabors in Ann Arbor (Universität Michigan) mit Kollegen vom Kinderkrankenhaus in Toronto [1]. Unterstützt von den NIH und der Cystischen Fibrose-Stiftung, konnte in jahrelanger, akribischer harter Arbeit die Funktion des Proteins aufgeklärt werden, das in der CF verändert ist und als cystic fibrosis transmembrane conductance regulator (CFTR) bezeichnet wird. Das CFTR-Protein ist ein Kanal in der Zellmembran, der das Salz- und Wassergleichgewicht in Lunge und anderen Teilen des Körpers reguliert. Abbildung 2.
 Abbildung 2. Das CFTR-Protein. Kryoelektronenmikroskopie. Das aus 1480 Aminosäuren bestehende Protein fungiert als Kanal für Chloridionen in den Membranen von Zellen, die Mukus, Schweiß , Speichel, Tränen und Verdauungsenzyme produzieren. Die Pore des trichterförmigen Kanals wird durch das graue Gitter angezeigt. (Beschriftung von der Redaktion eingefügt; Bild: Credit: Zhang & Chen, 2016, Cell 167, 1586–1597.)
Abbildung 2. Das CFTR-Protein. Kryoelektronenmikroskopie. Das aus 1480 Aminosäuren bestehende Protein fungiert als Kanal für Chloridionen in den Membranen von Zellen, die Mukus, Schweiß , Speichel, Tränen und Verdauungsenzyme produzieren. Die Pore des trichterförmigen Kanals wird durch das graue Gitter angezeigt. (Beschriftung von der Redaktion eingefügt; Bild: Credit: Zhang & Chen, 2016, Cell 167, 1586–1597.)
Neue Technologien wie die Kryo-Elektronenmikroskopie (das Journal Nature hat diese zur Methode des Jahres 2015 gekürt) gaben Forschern erst in jüngster Zeit die Möglichkeit die genaue Struktur des Proteins mit den Mutationen zu kartieren, die zu CF führen.
An cystischer Fibrose erkrankte Menschen tragen eine Mutation in beiden Kopien des CFTR-Gen - d.i. sowohl in der vom Vater als auch in der von der Mutter vererbten Kopie. Abbildung 3.
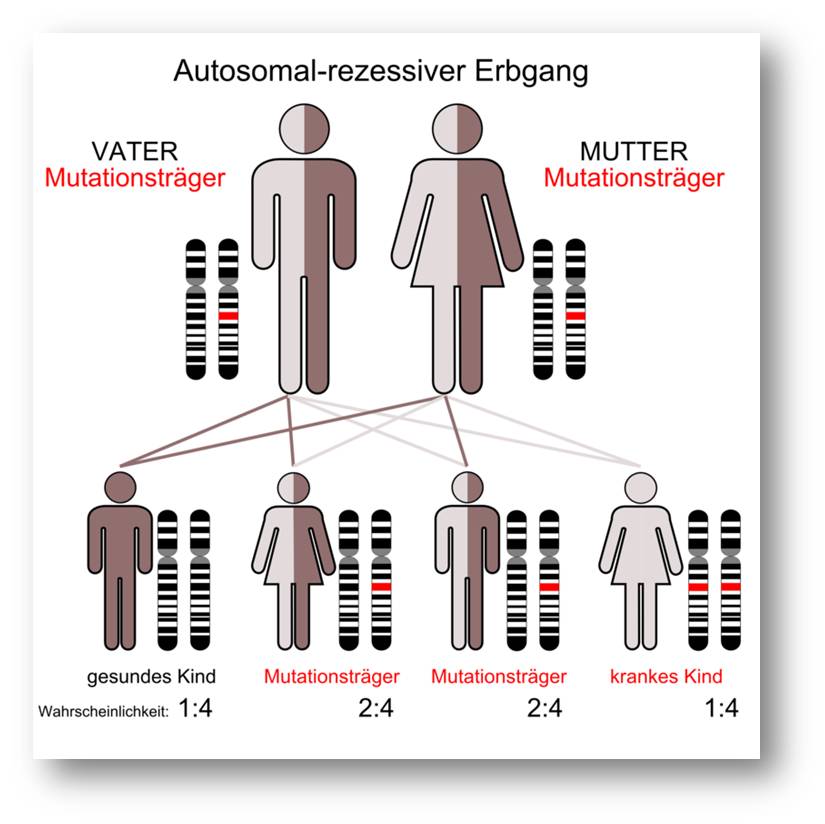 Abbildung 3. Cystische Fibrose ist bei uns die häufigste Erbkrankheit, jeder 20. Mensch trägt ein mutiertes CFTR-Gen. An CF-Erkrankte tragen Mutationen in beiden Kopien des Gens. (Bild von der Redaktion eingefügt; Quelle Armin Kübelbeck / Wikimedia.org. Lizenz: cc-by-sa)
Abbildung 3. Cystische Fibrose ist bei uns die häufigste Erbkrankheit, jeder 20. Mensch trägt ein mutiertes CFTR-Gen. An CF-Erkrankte tragen Mutationen in beiden Kopien des Gens. (Bild von der Redaktion eingefügt; Quelle Armin Kübelbeck / Wikimedia.org. Lizenz: cc-by-sa)
Bis jetzt sind mehr als 1700 derartige Mutationen bekannt, die CF auslösen können. Die häufigste Mutation - die sogenannte F508del Variante (in Position 508 des 1480 Aminosäuren langen Proteins fehlt die Aminosäure Phenylalanin - davon sind rund 2/3 der CF-Patienten betroffen; Anm. Red.) - resultiert in einem falsch gefalteten Protein, das abgebaut wird, bevor es noch seine richtige Position in der Zellmembran erreicht hat. Auf Grund des fehlenden Kanals wird nun der abgesonderte, die Zellen überlagernde dünnflüssige Mukus zu dickem, klebrigem Schleim verdichtet und kann u.a. zu lebensbedrohenden Infektionen und Lungenversagen führen.
Es war ein langer, beschwerlicher Weg Arzneimittel zu entwickeln, welche die Funktion von mutiertem CFTR restaurieren können.
Anfangs haben viele von uns gedacht, dass Gentherapie für die Behandlung dieser Erkrankung die Methode der Wahl wäre. Allerdings waren die Schwierigkeiten durch Gen-Transfer eine langanhaltende Korrektur des CF-Defekts in den Luftwegen zu erreichen ungemein entmutigend.
Aufbauend auf dem, aus NIH-unterstützter akademischer Forschung zunehmend vertieften Verständnis der CFTR-Funktion, entstand vor rund 20 Jahren eine Partnerschaft der Cystischen Fibrose-Stiftung und einer kleinen Firma, die sich Aurora nannte (und später zu Vertex Pharmaceuticals Inc., Boston wurde). Diese begann nach kleinen Molekülen zu suchen, welche einem anormalen CFTR-Protein zur korrekten Faltung verhelfen könnten - derartige Substanzen werden als Korrektoren bezeichnet - und solchen, welche die richtige Funktion ermöglichten, wenn das Protein dann die Zellmembran erreichte - derartige Substanzen werden als "Potentiatoren" (Verstärker) bezeichnet.
Rund 30 000 Amerikaner leiden an CF. Der erste größere Fortschritt in der Behandlung mit auf Zielmoleküle zugeschnittenen Arzneistoffen kam 2012, als die Food and Drug Administration (FDA) Ivacaftor (Kalydeco) zugelassen hat. So aufregend dies auch war, so wussten wir, dass es nur den ersten Schritt auf einem schwierigen Weg allen CF-Patienten zu helfen bedeutete. Dies ist der Fall, weil Kalydeco nur bei rund 4 % der Patienten wirkt, welche eine G551D Mutation tragen (an der Position 551 wurde ein Glycin durch eine Asparaginsäure ersetzt) zusammen mit anderen, die eine von 23 relativ seltenen Mutationen haben, die zu einem teilweise funktionsfähigen CFTR führen.
Der nächste bedeutende Meilenstein in der CF Behandlung wurde dann 2015 mit der Zulassung von Orkambi durch die FDA erreicht. Orkambi, eine Kombination von Ivacaftor mit Lumacaftor, dem ersten "Korrektor", wurde für Patienten bestimmt, die zwei Kopien der F508del Mutante besitzen. Für diese Patientengruppe liegt eine weitere Kombinationstherapie (Ivacaftor plus Tezacaftor) zur Prüfung bei der FDA.
Anlass zum Optimismus
bieten vorläufige Ergebnisse aus klinischen Studien, die vergangene Woche veröffentlicht wurden [2].
Es wurden hier in der Klinik (in Phase I und II) drei "Next-Generation" Dreifachkombinationstherapien untersucht, welche die Funktion des CFTR modulieren. Diese Strategie geht tatsächlich bei 90 % der Patienten, die zumindest eine Kopie von F508del tragen, auf. Vertex berichtete am Dienstag der letzten Woche:
- mit allen drei "Next-Generation" Dreifachkombinationstherapien konnten markante Verbesserungen der Lungenfunktion bei Patienten erzielt werden, die eine F508del Mutation und eine Minimal-Funktions-Mutante trugen - dies war bis dahin eine Form der CF, die besonders schwierig zu behandeln war. Konkret bedeutete dies: auf eine zwei- bis vierwöchige Behandlung mit der Kombination aus zwei Korrektoren und einem Verstärker sprachen die Patienten mit 10 Prozent Verbesserung des forcierten expiratorischen Volumens pro Sekunde (FEV1) an, welches die zentrale Größe für die Lungenkapazität darstellt. Obwohl die Untersuchungen als Doppel-Blind-Studien ausgeführt wurden, fanden viele von den Patienten, welche die aktive Dreierkombination erhielten, schnell heraus, dass etwas neues, wundervolles mit ihnen passierte.
- Darüber hinaus: Die "Next-Generation" Dreifachkombinationstherapien waren generell gut verträglich und verringerten die Konzentrationen von Chlorid im Schweiß. Erhöhtes Chlorid im Schweiß ist über Jahrzehnte als diagnostischer Test für CF benutzt worden, der Nachweis einer Senkung ist ein starker Indikator dafür, dass die Arzneistoffe im gesamten Körper wirksam wurden.
Diese Ergebnisse werden - zusammen mit Daten aus einer weiteren klinischen Studie, die heuer beginnen soll, - für Vertex die Grundlage bilden, um die Substanz oder die Kombination von Substanzen auszuwählen, die in der ausgedehnteren Phase 3 Studie eingesetzt werden soll.
Alles in allem
ist nun für rund 40 % der an CF leidenden Menschen eine zielgerichtete Behandlung zugelassen. Wenn allerdings die neuen Dreifach-Kombinationen halten, was sie in den ersten Studien versprechen, so könnte nach Meinung von Michael Boyle, dem Alt-Vizepräsidenten der Cystic Fibrosis Foundation, Bethesda, es möglich werden, bis zu 90 % der an CF Erkrankten zu behandeln.
Unter den Zehntausenden CF-Patienten, denen die nächste Generation zielgerichteter Medikamente nützen sollte, ist auch die kleine Avalyn Mahoney aus Cardiff, CA, die eben zwei Jahre alt wurde. Noch vor wenigen Jahrzehnten hätten Kinder wie Avalyn das Teenageralter nicht überlebt. Dank der Fortschritte, die auf NIH-unterstützter Grundlagenforschung aufbauen, sind die Aussichten für sie und so viele andere wesentlich günstiger.
Das ist eine grandiose Nachricht. Dazu möchte ich anfügen, dass weder die Cystische-Fibrose-Stiftung noch die NIH ruhen werden, bis es effektive und leistbare Behandlungen - oder noch besser Heilungen - für jeden CF-Patienten in den US und auf dem gesamten Erdball geben wird.
[1] Identification of the cystic fibrosis gene: chromosome walking and jumping. Rommens JM1, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et al. Science. 1989 Sep 8;245(4922):1059-1065.
[2] Positive Early Study Results for Next-Generation CFTR Modulators. Cystic Fibrosis Foundation News Release, July 18, 2017. https://www.cff.org/News/News-Archive/2017/Positive-Early-Study-Results-...
* Dieser Artikel von NIH Director Francis Collins, M.D., Ph.D. erschien unter dem Titel:"Another Milestone in the Cystic Fibrosis Journey" zuerst (am 20. Juli 2017) im NIH Director’s Blog: https://directorsblog.nih.gov/2017/07/20/another-milestone-in-the-cystic... Der Artikel wurde geringfügig für den Blog adaptiert und einige Abbildungen wurden von der Redaktion eingefügt. Reprinted (and translated by ScienceBlog) with permission from the National Institutes of Health (NIH)..
Weiterführende Links
What is Cystic Fibrosis (National Heart, Lung, and Blood Institute/NIH).
Genetics Home Reference: Cystic Fibrosis (National Library of Medicine/NIH).
Krankheitsbild Mukoviszidose. Beschreibung und Video 2:32 min (deutsch)
Cystic fibrosis. Video (englisch) 8:28 min. Author: Osmosis(31 August 2016). License: cc-by-sa.
- Printer-friendly version
- Log in to post comments