Do, 02.03.2017 - 09:02 — Eva Maria Murauer

![]()
Die Schmetterlingskrankheit - Epidermolysis bullosa (EB) - ist eine derzeit (noch) nicht heilbare, seltene Erkrankung, die durch Mutationen in Strukturproteinen der Haut hervorgerufen wird und in Folge durch eine extrem verletzliche Haut charakterisiert ist. Dr. Eva Maria Murauer vom EB-Haus Austria zeigt, dass sich derartige Mutationen in den Stammzellen von Patienten mittels Gentherapie korrigieren lassen und aus den so korrigierten Zellen Hautäquivalente produziert werden können, welche die Haut von EB-Patienten stückweise ersetzen und (langfristig) die Charakteristik einer stabilen, gesunden Haut bewahren können.*
Was ist Epidermolysis bullosa (Schmetterlingskrankheit)?
Unter die Bezeichnung Epidermolysis bullosa (EB) fällt eine Reihe von angeborenen Erkrankungen, die durch eine extrem verletzliche Haut charakterisiert sind, welche bereits nach leichten mechanischen Reizen Blasen bildet, die Wunden nach sich ziehen. Da die Haut von EB-Patienten in ihrer Fragilität einem Schmetterlingsflügel gleicht, wird EB volkstümlich auch als Schmetterlingskrankheit bezeichnet und die (vorwiegend jungen) Patienten als "Schmetterlingskinder" (Abbildung 1).
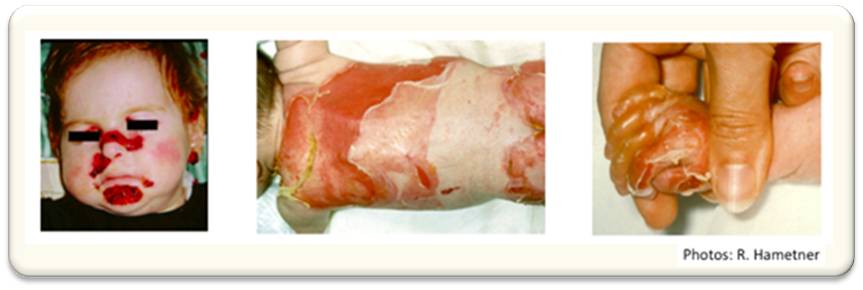 Abbildung 1. Epidermolysis bullosa (EB) ist eine angeborene, derzeit noch nicht heilbare Erkrankung, die nicht nur auf die Haut beschränkt bleibt. Bei geringster mechanischer Belastung bilden sich Blasen, es entstehen schmerzende Wunden, oft gefolgt von Vernarbungen. Bei manchen EB-Formen kommt es auch zu Verwachsungen von Fingern und Zehen.
Abbildung 1. Epidermolysis bullosa (EB) ist eine angeborene, derzeit noch nicht heilbare Erkrankung, die nicht nur auf die Haut beschränkt bleibt. Bei geringster mechanischer Belastung bilden sich Blasen, es entstehen schmerzende Wunden, oft gefolgt von Vernarbungen. Bei manchen EB-Formen kommt es auch zu Verwachsungen von Fingern und Zehen.
Allerdings bleibt die Erkrankung nicht auf die Haut beschränkt, es können auch innere Organe und die Schleimhäute (z.B. im Mund oder im Verdauungstrakt) betroffen sein, einige bullöse Erkrankungstypen führen zu sehr aggressiven Hautkrebsformen.
EB gehört zu den seltenen Erkrankungen, an der weltweit rund 500 000 Menschen- also einer von 17 000 - leiden (in Österreich rechnet man mit rund 300 "Schmetterlingskindern"). Die Ursachen von EB sind bekannt: es wurden bis jetzt Mutationen in mehr als 18 Genen identifiziert, die für Strukturproteine in der Haut kodieren, welche die Haftung der obersten Hautschichten - der Epidermis - an die darunterliegende Schicht - der Dermis - bewirken (Abbildung 2). Ist eines dieser Proteine defekt oder nicht mehr (in ausreichender Menge) vorhanden, so ist der Zusammenhalt der Hautschichten gestört und es kommt dort bereits bei leichten mechanischen Reizen zu Blasenbildung und schmerzenden Wunden. Je nachdem welches Protein an welcher Stelle der Epidermis/Dermis defekt ist, resultieren unterschiedlich schwer verlaufende Typen von EB (Abbildung 2).
 Abbildung 2. Epidermolysis bullosa wird durch defekte und/oder fehlende Strukturproteine (rechts) verursacht, die für den Zusammenhalt der Hautschichten Epidermis und Dermis (linkes Schema und Histologie, Mitte) essentiell sind. Je nachdem wo die Blasenbildung auftritt werden unterschiedliche EB-Typen definiert (EBS: EB simplex, JEB: junktionale EB, DEB: dystrophe EB, KS: Kindler Syndrom).
Abbildung 2. Epidermolysis bullosa wird durch defekte und/oder fehlende Strukturproteine (rechts) verursacht, die für den Zusammenhalt der Hautschichten Epidermis und Dermis (linkes Schema und Histologie, Mitte) essentiell sind. Je nachdem wo die Blasenbildung auftritt werden unterschiedliche EB-Typen definiert (EBS: EB simplex, JEB: junktionale EB, DEB: dystrophe EB, KS: Kindler Syndrom).
Wie kann man EB behandeln?
EB ist derzeit (noch) nicht heilbar, es können bloß Symptome behandelt werden, in langwierigen Prozeduren Wunden verbunden, Infektionen vorgebeugt und Schmerzen gelindert werden. Öffentliche Gesundheitssysteme sind auf die besonderen Herausforderungen einer derartigen seltenen Erkrankung kaum vorbereitet. Vor 22 Jahren wurde in Großbritannien von den Eltern an EB erkrankter Kinder und Ärzten eine Selbsthilfegruppe - DEBRA - gegründet. Inzwischen ist DEBRA zu einem internationalen Netzwerk in mehr als 50 Ländern angewachsen und seit 1995 auch in Österreich präsent. Die Organisation finanziert sich aus Spenden. Ihr Ziel ist es die Lebensqualität der Betroffenen zu verbessern, kompetente medizinische Versorgung anzubieten und durch weltweite Förderung von Spitzenforschung mitzuhelfen, wirksame Therapien zu finden und zu entwickeln.
An Strategien zur Behandlung dieser Erkrankungen wird intensiv geforscht - im Fokus stehen vor allem Strategien zur Gentherapie, die ja die Ursache der Erkrankung beheben könnte, daneben das Einbringen von Zellen gesunder Spender (Zelltherapie), das Ersetzen defekter Strukturproteine und die medikamentöse Behandlung zur Linderung der Beschwerden (Abbildung 3).
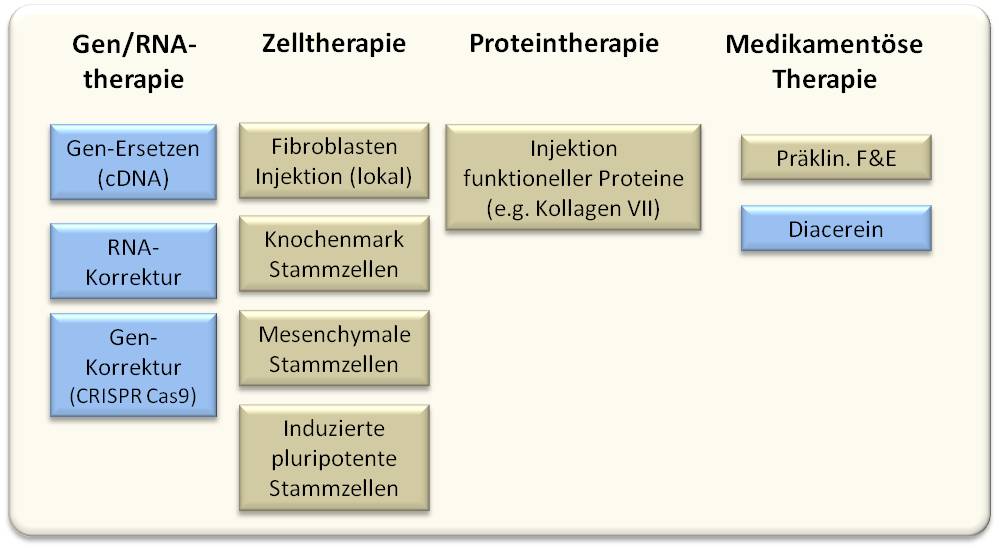 Abbildung 3. Strategien zur Therapie von Epidermolysis bullosa. Die blau hervorgehobenen Kästchen zeigen einige der von der Forschungseinheit im EB-Haus Austria verfolgten Strategien. Diacerein ist ein synthetisches anti-entzündliches Arzneimittel, das derzeit für EB in der klinischen Entwicklung ist.
Abbildung 3. Strategien zur Therapie von Epidermolysis bullosa. Die blau hervorgehobenen Kästchen zeigen einige der von der Forschungseinheit im EB-Haus Austria verfolgten Strategien. Diacerein ist ein synthetisches anti-entzündliches Arzneimittel, das derzeit für EB in der klinischen Entwicklung ist.
Das EB-Haus in Salzburg
DEBRA Austria hat im Jahr 2005 eine Spezialklinik, das EB-Haus Austria, an den Salzburger Landeskliniken eröffnet, das mittlerweile im Bereich EB zu einem europaweit anerkannten "Centre of Expertise" geworden ist. Das EB-Haus ist Teil der Salzburger Universitätsklinik und ermöglicht so unter einem Dach eine umfassende Palette an Untersuchungen und Maßnahmen: von Diagnostik und medizinischer Versorgung für "Schmetterlingskinder" über Grundlagenforschung und klinische Studien bis hin zu Vernetzung und Ausbildung von medizinischem Fachpersonal und Betroffenen.
Im EB-Haus streben wir vor allem an, mittels Gentherapie ein defektes Gen durch ein intaktes zu ersetzen. Daneben entwickeln wir Methoden, um Mutationen auf der Ebene der RNA-Moleküle zu korrigieren - das sogenannte mRNA trans-splicing -, eine Methode, in der kein ganzes Gen eingebracht wird, sondern nur der defekte Teil eines Gens ausgetauscht wird. Seit kurzem untersuchen wir auch den Einsatz einer Genkorrektur mittels der CRISPR-Cas9 Genschere. (Abbildung 3.) Alle diese Methoden bieten viele Vorteile, haben aber auch Schwachstellen.
Eine erfolgreiche ex-vivo Gentherapie - eine Fallstudie
Primär beschäftigen wir uns mit der Entwicklung einer ex-vivo Gentherapie. Dabei werden dem Patienten Hautproben entnommen und die darin befindlichen Stammzellen isoliert. In diese Stammzellen wird dann in der Zellkultur das intakte Gen eingefügt, die so korrigierten Zellen vermehrt und zu sogenannten "skin sheets" (dünnen Oberhautschichten) gezüchtet. Mit dieser neuen Haut können dann Wundflächen des Patienten abgedeckt werden. Da diese Haut einen gesunden Ersatz für das früher defekte Strukturprotein produziert, ist der Zusammenhalt der Hautschichten wieder hergestellt, daher sollte es hier keine Blasenbildung mehr geben. Dies haben wir vor kurzem an einer EB-Patientin verifizieren können.
Es handelte sich dabei um eine vom österreichischen Gesundheitsministerium genehmigte Studie, die in Zusammenarbeit mit dem Zentrum für Regenerative Medizin in Modena (Italien) erfolgte. Die einzelnen Schritte der ex-vivo Gentherapie sind in Abbildung 4 schematisch dargestellt. Die Patientin, eine 49-jährige Frau, litt an der junktionalen EB-Form: sie hatte ein defektes Laminin-Gen und konnte damit das Laminin Protein, einen wesentlichen Bestandteil der Basalmembran zwischen Epidermis und Dermis, nicht bilden. Seit Geburt litt sie an schweren Hauterosionen. Insbesondere hatte sie seit mehr als 10 Jahren eine große offene Wunde am rechten Bein, die trotz aller Therapieversuche nicht heilte und laufend zu schweren Infektionen führte.
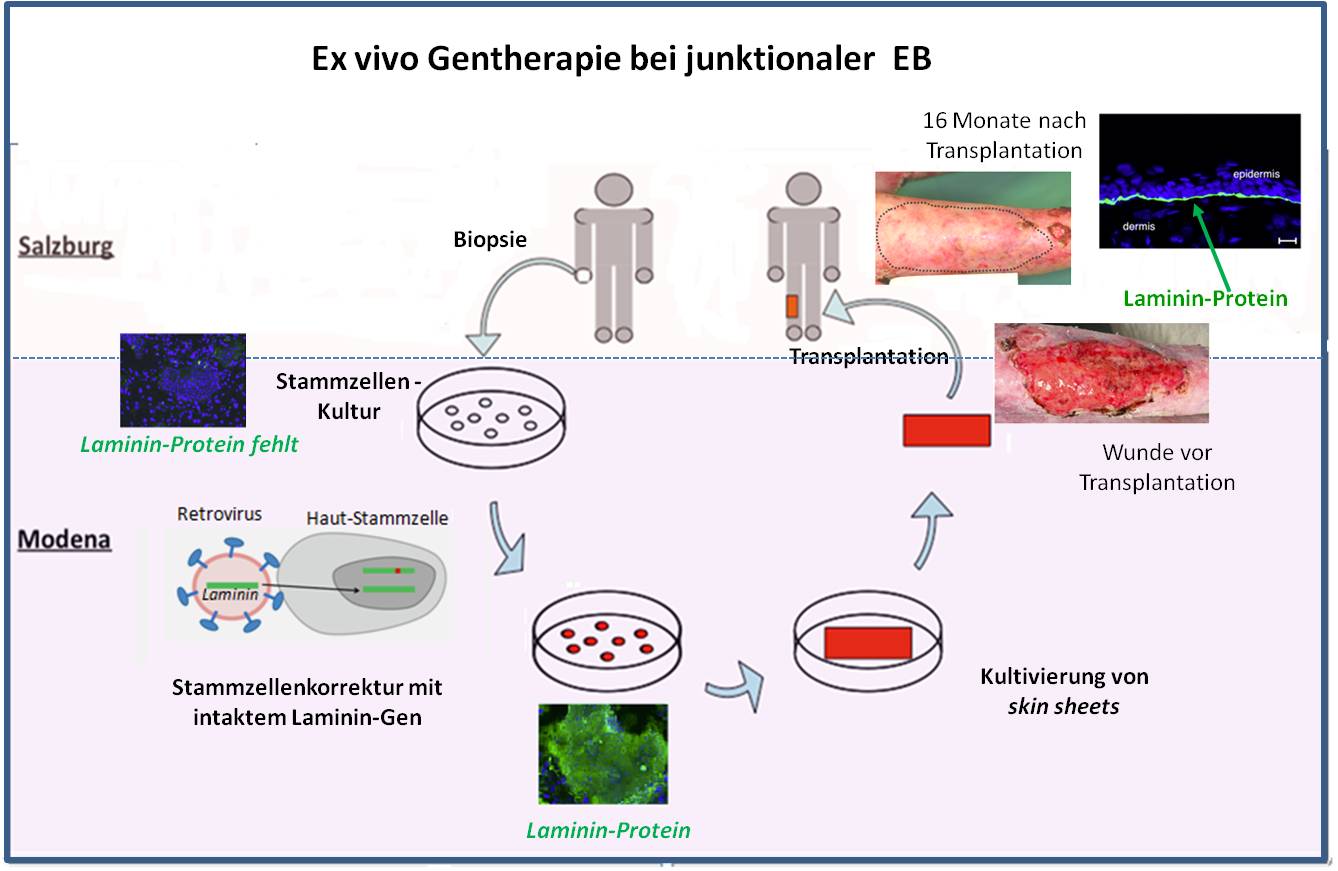 Abbildung 4. Erfolgreiche Heilung einer Beinwunde bei junktionaler EB durch Transplantation von körpereigenen korrigierten Stammzellen der Haut. Kooperation: EB-Haus Austria (Salzburg) und Zentrum für Regenerative Medizin, Modena (Italien). Das defekte Laminin-Gen wurde durch ein intaktes Gen ersetzt. Immunfluoreszenzfärbung (grün) zeigt das Laminin-Protein in den korrigierten Stammzellen und 1 Jahr nach Transplantation in einer Hautbiopsie an der korrekten Position zwischen Epidermis und Dermis. Außerhalb der transplantierten Fläche (schwarz umrandet) treten weiter Erosionen auf. (Die obersten 3 Fotos stammen aus J.Bauer et al., 2016 [1] und sind unter cc-by-sa-nd lizensiert.)
Abbildung 4. Erfolgreiche Heilung einer Beinwunde bei junktionaler EB durch Transplantation von körpereigenen korrigierten Stammzellen der Haut. Kooperation: EB-Haus Austria (Salzburg) und Zentrum für Regenerative Medizin, Modena (Italien). Das defekte Laminin-Gen wurde durch ein intaktes Gen ersetzt. Immunfluoreszenzfärbung (grün) zeigt das Laminin-Protein in den korrigierten Stammzellen und 1 Jahr nach Transplantation in einer Hautbiopsie an der korrekten Position zwischen Epidermis und Dermis. Außerhalb der transplantierten Fläche (schwarz umrandet) treten weiter Erosionen auf. (Die obersten 3 Fotos stammen aus J.Bauer et al., 2016 [1] und sind unter cc-by-sa-nd lizensiert.)
Von dieser Patientin entnahmen wir Hautbiopsien aus der Handfläche. Das Team in Modena isolierte daraus Hautstammzellen und korrigierte diese mit einem intakten Laminin-Gen. Dabei wurde ein Retrovirus als Vektor verwendet, der das Gen stabil in die Zellen integrierte. Als wir die korrigierten Zellen untersuchten, zeigte es sich, dass alle Zellen nun ein intaktes Laminin (Abbildung 4, ganz unten) produzierten: Dies war nun keine Korrektur des mutierten Gens - dieses befand sich weiter in den Zellen, es war bloß ein zusätzliches intaktes Gen zugefügt worden (Abbildung 4, Schema).
Aus den so korrigierten Stammzellen wurden dann zwei skin sheets im Ausmaß von jeweils 35 cm2 produziert. Diese neue Haut transplantierten wir dann auf die Beinwunde der Patientin. Bereits neun Tage danach konnten wir dort regenerierende Haut beobachten, Nachuntersuchungen auch nach 12 Monaten zeigten stabile, intakte Haut, die auch nach mechanischem Reiben keine Blasen mehr bildete. Um nachzuweisen, dass diese Haut noch korrigierte Zellen enthielt, haben wir Gewebeproben entnommen und histologisch auf das Vorhandensein des Laminin-Proteins geprüft. Dies war der Fall: Das eingebrachte Laminin-Gen produzierte an der Grenzfläche von Epidermis/Dermis in den Hauttransplantaten nachhaltig funktionierendes Laminin-Protein (Abbildung 4, rechts oben).
Unerwünschte Nebeneffekte der Behandlung traten bis jetzt nicht auf.
Diesen erfolgreichen Ansatz wollen wir nun an einer weiteren EB-Form - der dystrophen EB - in einer Phase 1/Phase 2 klinischen Studie prüfen. Bei dieser EB-Form fehlt den Patienten zwischen Epidermis und Dermis das Protein Kollagen VII, dies führt zu sehr schweren Blasenbildungen. Die Studie wurde von den zuständigen österreichischen Behörden sowie der Ethikkommission bereits genehmigt und kann an bis zu zwölf Patienten durchgeführt werden.
Erste Versuche zur Korrektur des Kollagen VII-Gens mittels der CRISPR Cas9 Technologie
Bei dieser neuen Technologie wird kein zusätzliches Gen in die Hautzelle eingebracht, sondern die Mutation wird im defekten Gen direkt und bleibend korrigiert. Sind dominant vererbte Erkrankungen durch ein falsch funktionierendes Protein bedingt, so ist dies zweifellos gegenüber der oben beschriebenen Methode von Vorteil - es wird kein zusätzliches, fehlerhaftes Protein mehr produziert. Ein weiterer wichtiger Vorteil besteht darin, dass kein Virusvektor notwendig ist, um die Genreparatur zu bewerkstelligen und damit kein, wenn auch geringes, Risiko einer Tumorentstehung eingegangen wird.
Wir haben nun die CRISPR Cas9 Technologie benützt, um eine Mutation auf dem Kollagen VII-Gen in den Hautzellen eines Patienten zu korrigieren, der an dystropher EB leidet. Bei dieser Methode wird das Enzym Cas9 eingesetzt, um das Gen nahe der mutierten Stelle durchzuschneiden, wobei die präzise Positionierung der Schnittstelle durch ein an Cas9 gebundenes kurzes Gegenstück zur zu schneidenden DNA - einer "guide RNA" - ermöglicht wird. Wir haben ein derartiges Cas9-guide RNA-Konstrukt zusammen mit einer Donor-Sequenz (ohne Mutation) angewandt, welche den DNA Strangbruch reparierte ("homologe Rekombination") und zu einem korrekten Kollagen 7-Gen führen sollte. Dies war der Fall: Die korrigierten Zellen produzierten nun das Kollagen VII-Protein (Abbildung 5).
 Abbildung 5. Erfolgreiche Korrektur des mutierten Kollagen VII- Gens mittels der CRISPR Cas9 Technologie. (Das Schema von CRISPR Cas9 stammt aus: M.Jinek et al., 2013, https://elifesciences.org/content/2/e00471 und ist unter cc-by-sa lizensiert.)
Abbildung 5. Erfolgreiche Korrektur des mutierten Kollagen VII- Gens mittels der CRISPR Cas9 Technologie. (Das Schema von CRISPR Cas9 stammt aus: M.Jinek et al., 2013, https://elifesciences.org/content/2/e00471 und ist unter cc-by-sa lizensiert.)
In weiterer Folge untersuchten wir - im Tierversuch -, ob die korrigierten Zellen nun eine einwandfreie Haut bilden können, mit dem Kollagen VII-Protein an der richtigen Position zwischen Epidermis und Dermis. Dazu haben wir die Zellen in Kultur zu Hautäquivalenten wachsen lassen und diese dann auf den Rücken von Mäusen transplantiert (um die Abstoßung des humanen Gewebes zu verhindern, erfolgten die Versuche an immundefizienten Mäusen). Vier Wochen später wurden dann Biopsien der angewachsenen Transplantate untersucht. Tatsächlich fand sich Kollagen VII an der richtigen Position.
Fazit
EB verursachende Mutationen lassen sich prinzipiell in den Hautzellen von EB-Patienten mittels Gentherapie korrigieren. Die aus korrigierten Zellen produzierten Hautäquivalente bewahren (langfristig) die Charakteristik einer stabilen, gesunden Haut und könnten die Haut von EB-Patienten stückweise ersetzen. Primär geht es dabei um die Abdeckung besonders geschädigter Hautareale. Ähnliches gilt auch für die neue CRISPR Cas9 Technologie: erste Versuche zeigen, dass diese (relativ) einfache und risikoarme Methode zweifellos geeignet ist, um EB-verursachende Mutationen und den EB-Phänotyp zu korrigieren. Bevor dieses Verfahren allerdings am Menschen erprobt werden kann, muss seine Effizienz noch gesteigert und seine Spezifität analysiert werden.
[1] J W. Bauer, Josef Koller, Eva M. Murauer, et al.,(2016) Closure of a Large Chronic Wound through Transplantation of Gene-Corrected Epidermal Stem Cells J Invest Dermatol. 2016 Nov 10. http://www.jidonline.org/article/S0022-202X(16)32636-7/pdf (open access under CC BY-NC-ND license)
*Eine ausführlichere Darstellung des Themas findet sich in dem Vortrag "Gene therapy approaches for epidermolysis bullosa, den die Autorin im November 2016 anlässlich der Feier zur Verleihung des Wilhelm-Exner Preises gehalten hat: https://slideslive.com/38899167/gene-therapy-approaches-for-epidermolysis-bullosa . Video (englisch) 15:45 min .
Weiterführende Links
DEBRA International: http://www.debra-international.org/homepage.html
DEBRA Austria: http://www.debra-austria.org/startseite.html
EB-Haus an den Salzburger Landeskliniken: www.eb-haus.org
Videos
DEBRA Austria - So fühlt sich das Leben für ein "Schmetterlingskind" an. (2015) Video 7:26 min Standard-YouTube-Lizenz
So fühlt sich das Leben für ein Schmetterlingskind an (2011, DEBRAustria) Video 7:56 min. Standard-YouTube-Lizenz
EB-Haus: Spezialklinik für Schmetterlingskinder (2012, DEBRAustria) Video 4:31 min. Standard-YouTube-Lizenz
Das Leben eines Schmetterlingskindes, Teil 1 (DEBRA Südtirol) Video 6:00 min, und Fortsetzung. Standard-YouTube-Lizenz
Artikel zum Thema Gentherapie im ScienceBlog.at
Francis S. Collins, 02.02.2017: Finden und Ersetzen: Genchirurgie mittels CRISPR/Cas9 erscheint ein aussichtsreicher Weg zur Gentherapie
Artikel zum Thema Haut im ScienceBlog.at
I.Schuster, 17.07.2015: Unsere Haut – mehr als eine Hülle. Ein Überblick. http://scienceblog.at/unsere-haut.
- Printer-friendly version
- Log in to post comments