Fr, 03.05.2024 — Ricki Lewis

![]() Hinweise auf die Bekämpfung einer verheerenden Krankheit können sich aus der Identifizierung von Menschen ergeben, die Genvarianten - Mutationen - haben, die sie schützen, indem sie die Krankheit verlangsamen oder überhaupt das Risiko ihrer Entstehung verringern. Wenn wir verstehen, wie sie dies tun, können wir Behandlungsstrategien für die gesamte Patientenpopulation entwickeln. Die Genetikerin Ricki Lewis berichtet über seltene Varianten von drei bereits gut untersuchten Genen - APOE-Cristchurch, Reelin und Fibronektin -, die vererbte Formen der Alzheimer-Krankheit zu verzögern scheinen - und zwar um Jahrzehnte. *
Hinweise auf die Bekämpfung einer verheerenden Krankheit können sich aus der Identifizierung von Menschen ergeben, die Genvarianten - Mutationen - haben, die sie schützen, indem sie die Krankheit verlangsamen oder überhaupt das Risiko ihrer Entstehung verringern. Wenn wir verstehen, wie sie dies tun, können wir Behandlungsstrategien für die gesamte Patientenpopulation entwickeln. Die Genetikerin Ricki Lewis berichtet über seltene Varianten von drei bereits gut untersuchten Genen - APOE-Cristchurch, Reelin und Fibronektin -, die vererbte Formen der Alzheimer-Krankheit zu verzögern scheinen - und zwar um Jahrzehnte. *
Gen Nr. 1: Der berühmte Fall der aus einer kolumbianischen Familie stammenden Aliria
Im Jahr 2019 berichteten Forscher über die Patientin Aliria Rosa Piedrahita de Villegas, die dank einer Variante eines zweiten, offenbar schützenden Gens die früh einsetzende familiäre Alzheimer-Krankheit abgewehrt zu haben schien. Der Bericht erschien in Nature Medicine [1].
Aliria gehört zu einer 6 000 Mitglieder zählenden Familie in Kolumbien, die dafür bekannt ist, dass viele Menschen im Alter von etwa 44 Jahren Symptome von Alzheimer zeigen, die verdächtige Anhäufung von Amyloid-Beta-Protein aber bereits in ihren Zwanzigern auftrat. Etwa die Hälfte der Familie ist davon betroffen. Sie haben eine Variante des Presenilin-1-Gens (PSEN1 E280A), die für etwa 70 Prozent der Fälle von früh einsetzender Alzheimer-Krankheit verantwortlich ist. Aliria hat die Variante geerbt, doch bei ihr setzte der kognitive Abbau erst im Alter von 72 Jahren ein.
"Unsere Arbeit mit dieser Familie ermöglicht es uns, die frühesten Veränderungen, die mit der Alzheimer-Krankheit in Verbindung gebracht werden, zu verfolgen und festzustellen, wie diese Veränderungen im Laufe der Zeit ablaufen. Dies wird uns helfen, über die Menschen in Kolumbien hinaus festzustellen, wer gefährdet und wer resistenter gegen Alzheimer ist, und zu lernen, welche Biomarker das Fortschreiten der Krankheit besser vorhersagen können", erklärte Dr. Yakeel T. Quiroz vom Massachusetts General Hospital gegenüber der Alzheimer's Association. Dort wurden bei dieser speziellen Patientin Hirnscans durchgeführt, die die für die Krankheit charakteristischen sehr hohen Konzentrationen von Amyloid-Beta-Protein-Plaques aufzeigten.
Aber was genau hat Aliria geschützt?
Sie hatte auch zwei Kopien der Christchurch-Mutation geerbt, einer seltenen Variante desApolipoprotein E (APOE)-Gens, benannt nach dem Ort, an dem es in Neuseeland entdeckt wurde. Die Christchurch-Genvariante verringert die Dichte der Tau-Fibrillen (Tangles), der anderen Art von Protein, das im Alzheimer-Gehirn aggregiert. Abbildung 1.
|
|
|
Abbildung 1 Stliisierte Darstellung der Amyloid-beta-Plaques (braun) im extrazellulären Raum zwischen den Neuronen und den Tau-Protein Knäueln (blau) in den Neuronen. |
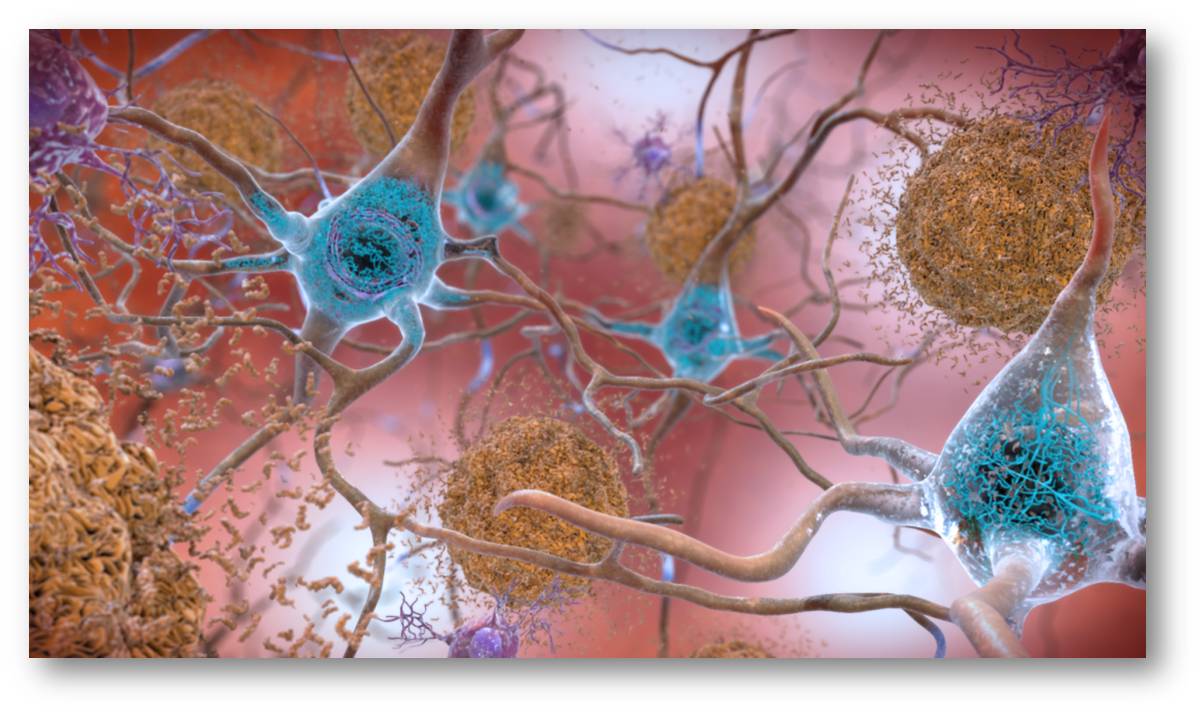
Kürzlich haben Forscher der Washington University School of Medicine mit genetisch veränderten "humanisierten" Mäusen gezeigt, dass die Christchurch-Mutation die Wechselwirkung zwischen Amyloid-Beta und Tau unterbindet.
"Wenn Menschen altern, beginnen viele eine Amyloid-Anhäufung im Gehirn zu entwickeln. Anfangs bleiben sie kognitiv unauffällig. Nach vielen Jahren jedoch beginnt die Amyloidablagerung zu einer Anhäufung des Tau-Proteins zu führen. Wenn dies geschieht, kommt es bald zu kognitiven Beeinträchtigungen. Wenn wir einen Weg finden, die Auswirkungen der APOE-Christchurch-Mutation zu imitieren, können wir vielleicht verhindern, dass Menschen, die bereits auf dem Weg zur Alzheimer-Demenz sind, diesen Weg weiter fortsetzen", erklärt Dr. David M. Holtzman von der Universität Washington.
Könnte auf therapeutischem Weg die Bildung von Amyloid-Beta-Plaques von der Ablagerung von Tau-Fibrillen entkoppelt werden?
Das war's, was die Forscher herausfinden wollten, die nun ihre Ergebnisse im Januar 2024 in der Zeitschrift Cell veröffentlichten [2]. Der Übergang von der Amyloidbildung zur Wechselwirkung mit Tau-Protein ist kritisch und noch wenig verstanden. Der Fall von Aliria könnte hier zur Klöärung beitragen.
"Der Fall dieser Frau war sehr, sehr ungewöhnlich, da sie die Amyloid-Pathologie aufwies, aber kaum eine Tau-Pathologie und nur sehr leichte kognitive Symptome, die erst spät auftraten. Das legte uns nahe, dass sie Hinweise auf die Verbindung zwischen Amyloid und Tau bieten könnte", so Holtzman.
Allerdings: Aliria ist ein Einzelfall; weltweit die einzige Person, von der man weiß, dass sie die beiden Mutationen aufweist. Haben diese tatsächlich zusammengewirkt, oder ist ihr ungewöhnlicher , offensichtlich vor Alzheimer schützender Genotyp nur ein Zufall?
Um dieses Rätsel zu lösen, setzte das Team um Holtzman genetisch manipulierte Mäuse ein, die übermäßig viel Amyloid produzierten und dazu auch die menschliche Christchurch-Mutation aufwiesen. Dann injizierten sie menschliches Tau-Protein in die Gehirne der Mäuse. Da die Gehirne der Mäuse bereits voller Amyloid-"Keime" waren, hätten sich die Tau-Knäuel an den Injektionsstellen festsetzen und dann auf andere Hirnregionen ausbreiten müssen - aber das taten sie nicht. Es wurde nur sehr wenig Tau inmitten des reichlich vorhandenen Amyloids gefunden, das die Gehirne erstickte - es war ein Modell für Alirias zufällige Doppelmutation.
Die Mäuse zeigten auf, wie die Christchurch-Mutation schützend wirkte, nämlich durch die Aktivität der Mikroglia, die als Abfallbeseitigungszellen des Gehirns fungieren. Mikroglia sammeln sich um Amyloid-Plaques auf Gehirnzellen. Bei Alzheimer-Mäusen mit der APOE-Christchurch-Mutation liefen die Mikroglia, die die Amyloid-Plaques umgaben, auf Hochtouren, um die Tau-Aggregate zu verschlingen und zu zerstören.
"Diese Mikroglia nehmen das Tau auf und bauen es ab, bevor sich die Tau-Pathologie wirksam auf die nächste Zelle ausbreiten kann. Ohne Tau-Pathologie kommt es nicht zu Neurodegeneration, Atrophie und kognitiven Problemen. Wenn wir die Wirkung der Mutation nachahmen können, könnten wir die Amyloid-Ansammlung unschädlich machen oder zumindest viel weniger schädlich machen und die Menschen vor der Entwicklung kognitiver Beeinträchtigungen schützen", so Holtzman.
Gen Nr. 2: Reelin
Die Forscher, die die große kolumbianische Familie untersuchten, zu der auch Aliria gehört, beschrieben in der Zeitschrift Nature Medicine vom 15. Mai 2023 "den weltweit zweiten Fall mit nachgewiesener extremer Resilienz gegenüber der autosomal dominanten Alzheimer-Krankheit". Wie Aliria behielt der Mann seine kognitiven Fähigkeiten bis zum Alter von 67 Jahren, obwohl er die starke PSEN1 E280A-Mutation hatte, die eine sehr früh einsetzende Alzheimer-Krankheit verursacht.
Im Alter von 73 Jahren zeigten Neuroimaging-Untersuchungen höhere Werte von Amyloid-Beta-Plaques als bei anderen Familienmitgliedern mit der Alzheimer-Mutation. Und obwohl sein Gehirn auch Tau-Knäuel aufwies, war der entorhinale Kortex - ein Gedächtniszentrum - vergleichsweise frei von Tau. Vielleicht ermöglichte diese ungewöhnliche Verteilung, dass seine kognitiven Fähigkeiten trotz des Ertränkens in Amyloid-Beta erhalten blieben.
Aber er hatte nicht die Christchurch-Mutation von Aliria.
Stattdessen wurde der Mann offenbar durch eine seltene Variante eines anderen Gens, RELN, geschützt, das für das Protein Reelin kodiert. Dabei handelt es sich um ein gut untersuchtes Signalprotein, das bei verschiedenen neurologischen und psychiatrischen Erkrankungen wie Schizophrenie, bipolarer Störung und Autismus-Spektrum-Störung eine Rolle spielt.
In Experimenten mit Mäusen zeigten die Forscher, dass diese Form von Reelin eine Gain-of-Function-Mutation ist, d.i. eine verbesserte Funktion im Vergleich zur häufigeren Variante des Gens besitzt. Wie Apolipoprotein E (APOE) bindet Reelin an Rezeptoren auf bestimmten Lipoproteinen, die Cholesterin transportieren. Dadurch wird die Aktivierung von Tau verringert, was offenbar das empfindliche Gleichgewicht zwischen Amyloid-Beta und Tau auf eine Weise stört, die die Alzheimer-Krankheit verlangsamt.
Gen Nr. 3: Veränderung von Fibronektin an der Blut-Hirn-Schranke
In jüngster Zeit haben sich Forscher der Columbia University auf eine andere, offenbar schützende Variante eins Gens konzentriert, das für das Protein Fibronektin kodiert. Es ist im gesunden Gehirn in nur sehr geringen Mengen vorhanden, eine hohe Konzentration wird aber mit einem erhöhten Risiko für die Entstehung von Alzheimer in Verbindung gebracht.
Fibronektin ist in die Blut-Hirn-Schranke eingebettet. Dieses 400 Meilen lange Labyrinth aus Kapillaren, den kleinsten Blutgefäßen, windet sich durch die Neuronen und Gliazellen, die die empfindliche Hirnsubstanz bilden.
Die aus einer einzelnen Zellschichte bestehenden Kapillarwände, das so genannte Endothel, bilden eine Auskleidung, die normalerweise so dicht gepackt ist, dass sie Giftstoffe aus dem Blutkreislauf fernhält, aber lebenswichtige Stoffe wie Sauerstoff ins Gehirn eintreten lässt. Die Schranke mildert auch biochemische Fluktuationen, die das Gehirn überfordern würden, wenn es ständig reagieren müsste, und überwacht den Gehalt an Neurotransmittern.
Zu verstehen, wie die Blut-Hirn-Schranke funktioniert, ist zentrales Thema der Arzneimittel-Forschung bei neurologischen Erkrankungen.
Aus Untersuchungen an Zebrafischen und Mausmodellen der Alzheimer-Krankheit haben die Forscher entdeckt, dass eine Variante des Fibronektin-Gens die Anhäufung von Fibronektin an der Blut-Hirn-Schranke verhindert. Überschüssiges Amyloid-Beta kann dann aus dem Gehirn in den Blutkreislauf eintreten, was das Risiko, an Alzheimer zu erkranken, verringert, so die Forscher. Ihre Forschungsergebnisse sind in Acta Neuropathologica [4] veröffentlicht.
Caghan Kizil, PhD, erklärt die Ergebnisse: "Die Alzheimer-Krankheit beginnt zwar mit Amyloid-Ablagerungen im Gehirn, aber die Krankheitsmanifestationen sind das Ergebnis von Veränderungen, die nach dem Auftreten der Ablagerungen stattfinden. Überschüssiges Fibronektin könnte die Beseitigung von Amyloidablagerungen im Gehirn verhindern. Unsere Ergebnisse deuten darauf hin, dass einige dieser Veränderungen in den Blutgefäßen des Gehirns stattfinden und dass wir in der Lage sein könnten, neue Arten von Therapien zu entwickeln, die die schützende Wirkung des Gens nachahmen, um die Krankheit zu verhindern oder zu behandeln."
Diese Strategie unterscheidet sich von einem direkten Angriff auf die Amyloid-Ablagerungen und einer verbesserten Beseitigung, die zu gering ausfallen und zu spät kommen dürften. "Wir müssen möglicherweise viel früher mit der Beseitigung von Amyloid beginnen, und wir glauben, dass dies über den Blutkreislauf geschehen kann. Deshalb freuen wir uns über die Entdeckung dieser Fibronektin-Variante, die ein gutes Target für die Entwicklung von Medikamenten sein könnte", so Richard Mayeux, Koautor der Studie.
Nachdem die Forscher die Folgen einer Senkung des Fibronektins in Zebrafischen und Mäusen nachgewiesen hatten, haben sie das Protein bei Menschen untersucht, die eine Variante des APOE-Gens, das mit Alzheimer assoziierte APOEε4 geerbt haben, aber lange leben. Hat eine Variante des Fibronektin-Gens sie geschützt?
Um das herauszufinden, haben die Columbia-Forscher die Genome von mehreren hundert Personen mit APOEε4 untersucht, die älter als 70 Jahre und unterschiedlicher Herkunft waren. Einige von ihnen waren bereits an Alzheimer erkankt.
|
|
|
Abbildung 2 Die funktionell inaktive Fibronektin1-Variante schützt vor Alzheimer - Schematische Darstellung. Links: Situation im "normalen" Gehirn. Mit Apolipoprotein Eε4 (APOEε4 ) und nur geringen Mengen an Fibronektin werden Amyloid-Aggregate (Plaques) effizient von Microglia-Zellen abgebaut und die entstandenen Produkte über die Blut-Hirn-Schranke eliminiert ("Clearance"). Mitte: Situation bei Alzheimer - Genotyp APOEε4 zusammen mit einem Zuviel an Fibronektin. Fibronektin lagert sich in der Blut-Hirnschranke an und führt zu deren Verdickung. Abbau von Plaques und Eliminierung von Amyloidprodukten brechen zusammen, Neuronen werden geschädigt ("Synaptic Degeneration"). Rechts: Die funktionell inaktive ("Loss of Function") Fibronektin-Variante lagert sich nicht an der Blut-Hirn-Schranke ab. Die Eliminierung von Amyloidprodukten kann erfolgen. Die Bildung von Amyloid-Plaques erfolgt in reduziertem Ausmaß/verzögert. (Bild von der Redaktion eingefügt aus: Bhattarai, P et al., [4], https://doi.org/10.1007/s00401-024-02721-1 und leicht modifiziert. Lizenz: cc-by.) |
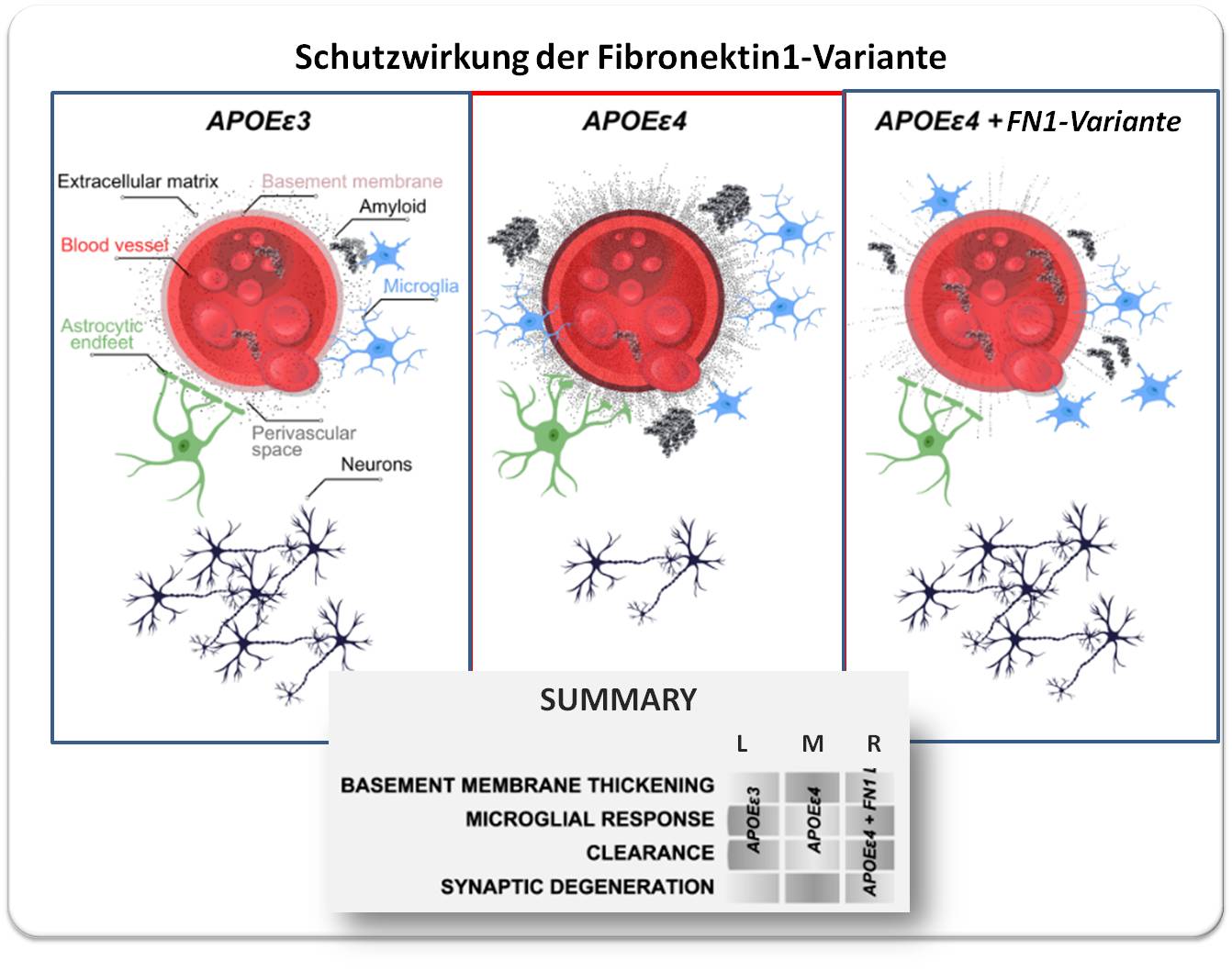
"Von den noch nicht erkrankten (resiliente)n Menschen können wir viel über die Krankheit erfahren und darüber, welche genetischen und nicht-genetischen Faktoren einen Schutz davor bieten könnten", sagt Badri N. Vardarajan, PhD, ein Coautor der Studie. Offensichtlich wirkt eine Variante des Fibronektin-Gens schützend.
Als die Forscher den Preprint ihrer Ergebnisse veröffentlichten, haben andere Teams Daten aus anderen Bevölkerungsgruppen hinzugefügt, die diesen Zusammenhang stützten: Fibronektin schützt. Die Daten von mehr als 11 000 Personen zeigten, dass die Mutation die Wahrscheinlichkeit, an Alzheimer zu erkranken, bei APOEε4 -Trägern um 71 % verringert und den Ausbruch der Krankheit bei denjenigen, die sie entwickeln, um etwa vier Jahre verzögert. Abbildung 2.
Nur etwa 1 bis 3 Prozent der APOEε4 -Träger in den USA besitzen auch die schützende Fibronektin-Mutation, das sind immerhin 200.000 bis 620.000 Menschen, schätzen die Forscher. Dieser angeborene Schutzschild könnte die Forschung inspirieren und zur Entwicklung neuer Medikamente führen, die noch viel mehr Menschen helfen könnten.
Kizil fasst zusammen: "Es gibt einen signifikanten Unterschied im Fibronektinspiegel in der Blut-Hirn-Schranke zwischen kognitiv gesunden Menschen und Alzheimer-Patienten, unabhängig von ihrem APOEε4 -Status. Alles, was überschüssiges Fibronektin reduziert, sollte einen gewissen Schutz bieten, und ein Medikament, das dies tut, könnte ein bedeutender Fortschritt im Kampf gegen diese zehrende Krankheit sein."
[1] Arboleda-Velasquez JF et al., Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. DOI: 10.1038/s41591-019-0611-3
[2] Chen Y et al., APOE3ch alters microglial response and suppresses Aβ-induced tau seeding and spread. Cell. 2024 Jan 18;187(2):428-445.e20. doi: 10.1016/j.cell.2023.11.029
[3] Lopera F. et al., Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat Med 29, 1243–1252 (2023). https://doi.org/10.1038/s41591-023-02318-3
[4] Bhattarai, P., Gunasekaran, T.I., Belloy, M.E. et al. Rare genetic variation in fibronectin 1 (FN1) protects against APOEε4 in Alzheimer’s disease. Acta Neuropathol 147, 70 (2024). https://doi.org/10.1007/s00401-024-02721-1.
* Der Artikel ist erstmals am 18. April 2024 in PLOS Blogs - DNA Science Blog unter dem Titel "Mutations in Three Genes Protect Against Alzheimer’s" https://dnascience.plos.org/2024/04/18/mutations-in-three-genes-protect-against-alzheimers/ erschienen und steht unter einer cc-by Lizenz. Die Autorin hat sich freundlicherweise mit der Übersetzung ihrer Artikel durch ScienceBlog.at einverstanden erklärt, welche so genau wie möglich der englischen Fassung folgt. Abbildung 2 plus Text wurden von der Redaktion aus der zitierten Publikation [4] eingefügt.
Im ScienceBlog: Zur Blut-Hirn-Schranke
Inge Schuster, 12.02.2024: Zur Drainage des Gehirngewebes über ein Netzwerk von Lymphgefäßen im Nasen-Rachenraum
Redaktion, 06.02.2020:Eine Schranke in unserem Gehirn stoppt das Eindringen von Medikamenten. Wie lässt sich diese Schranke überwinden?
Redaktion, 10.10.2017: Ein neues Kapitel in der Hirnforschung: das menschliche Gehirn kann Abfallprodukte über ein Lymphsystem entsorgen.
- Printer-friendly version
- Log in to post comments