Di, 15.10.2024— Inge Schuster

![]()
Die 2024 mit dem Nobelpreis in Chemie ausgezeichneten Wissenschafter Demis Hassabis, John Jumper und David Baker haben eine Revolution in der Proteinforschung ausgelöst. Hassabis und Jumper haben mit AlphaFold2 ein künstliches Intelligenzmodell entwickelt, das den Traum wahrmacht mit hoher Genauigkeit die räumliche Struktur eines Proteins aus seiner Aminosäuresequenz vorhersagen zu können. Baker ist es gelungen mit seinem kontinuierlich weiter entwickelten Computerprogramm Rosetta völlig neue, in der Natur nicht vorkommende Proteine mit speziellen Eigenschaften für diverse Anwendungen zu designen. AlphaFold2 und Rosetta sind offentlich frei zugänglich, ihre bereits millionenfache Nutzung führt in eine neue Ära von Grundlagenforschung und diversesten Anwendungen.
Vorweg ein kurzer Kommentar
Die Verleihung des Chemie-Nobelpreises [1. 2] an David Baker, Demis Hassabis und John Jumper kam keineswegs unerwartet: die ausgezeichneten Arbeiten sind doch wohl einer "major transition" (d.i. einem "großen Übergang" [3]) in der Möglichkeit biologische Systeme zu beschreiben/zu verstehen gleichzusetzen. Möglich gemacht wurde die radikale Neuerung durch reichlich vorhandene, günstige Ressourcen - einer enorm gestiegenen Leistungsfähigkeit der Rechner, die das Analysieren und Speichern von Big Data erlaubt und dem Zugriff auf Datenbanken, in denen das Ergebnis von mehr als 60 Jahren Forschung zu Proteinstrukturen für jedermann frei verfügbar ist. Die nun ausgezeichneten Computer-und KI-gestützten Technologien wurden in den wenigen Jahren seit ihrer Freigabe bereits von Millionen Forschern für "unzählige" Fragestellungen des Proteindesigns und der Strukturvorhersage genutzt. Die Ergebnisse werden wohl unsere Welt verändern.
Über Proteine,....
“The most significant thing about proteins is that they can do almost anything.” Francis Crick ,1958
Mit Proteinen und durch Proteine entstehen und vergehen alle Strukturen der belebten Materie. Proteine bilden Gerüste innerhalb und außerhalb der Zellen, binden andere Moleküle, transportieren diese und setzen sie um. Proteine ermöglichen den Informationsaustausch in und zwischen Zellen und ihrer Umgebung, fungieren als präzise Katalysatoren (Enzyme) der Stoffwechselvorgänge, synthetisieren und metabolisieren andere Proteine und bauen Fremdstoffe ab. Das Ablesen der in der DNA gespeicherten genetischen Information und deren Übersetzung in Proteine wird durch Proteinkomplexe gesteuert, das Hormon-, Immun- und neuronale System durch Proteine reguliert.
Proteine sind groß, Makromoleküle, die aus linearen Ketten von (über Peptidbindungen) miteinander verknüpften Aminosäuren aufgebaut sind. Diese bereits zu Beginn des 20. Jahrhunderts aufgestellte "Peptidhypothese" wurde 1951 bestätigt, als es Frederick Sanger und Hans Tuppy gelang die chemische Struktur des ersten Proteins, des aus 51 Aminosäuren bestehenden Hormons Insulin, aufzuklären (zu sequenzieren). Etwa in diese Zeit fallen auch die mit Hilfe der Röntgenstrukturanalyse aufgeklärte DNA-Struktur und die ersten erfolgreichen Versuche die räumliche Struktur von Proteinen zu ermitteln: Der aus Wien stammende Chemiker Max Perutz benötigte fast zwei Jahrzehnte, um das aus 4 Untereinheiten bestehende Hämoglobin zu analysieren, der Brite John Kendrew konnte, aufbauend auf den Erkenntnissen von Max Perutz, die Struktur des einfacher aufgebauten. kleineren Myoglobin in "nur" 11 Jahren lösen [4]. Aus den Strukturen wurde die Funktion dieser Proteine klar erkennbar, wie sich die Aminosäureketten falteten, wie und wo sie Sauerstoff banden und die Strukturen sich dabei veränderten. Wie 1958 schon Sanger wurden Perutz und Kendrew für diese Pionierleistungen 1962 mit dem Nobelpreis für Chemie ausgezeichnet.
........ die weitere experimentelle Ermittlung ihrer 3D-Strukturen........
Die Ergebnisse an Myoglobin und Hämoglobin zeigten klar: Um die Funktionen von Proteinen zu verstehen und gegebenenfalls manipulieren zu können, ist eine detaillierte Kenntnis ihrer räumlichen Struktur, ihrer Gestalt und ihrer physikalisch-chemischen Eigenschaften an Oberflächen und Domänen, an denen sie mit Bindungsartnern interagieren, notwendig.
In der Folgezeit nahmen mehr und mehr Forscher die lange Zeit noch sehr schwierigen, kosten- und arbeitsintensiven Bemühungen auf sich, um Proteine in der für die Strukturanalyse benötigten Menge zu isolieren, zu reinigen und zu kristallisieren. In der ersten, 1971 gegründeten öffentlich frei zugänglichen Proteindatenbank PDB (https://www.rcsb.org/pages/about-us/index) waren 1976 bereits 13 Strukturen eingetragen - alles einfach zu handhabende, sehr häufig vorkommende Proteine, darunter einige Protein-spaltende Enzyme (Proteasen) und die aus 4 Untereinheiten zusammengesetzte Laktatdehydrogenase [5]. In den 1980er-Jahren gelang es die Strukturen erster Membranproteine aufzuklären. Neue Methoden der Molekularbiologie, laufend verbesserte Techniken der Kristallisierung und Fortschritte in der Kristallstrukturanalyse verkürzten die Prozeduren und ließen ab den 1990-er Jahren die Zahl der neuen Strukturen sehr schnell anwachsen. Bei einem Zuwachs von jeweils mehr als 14 000 neuer Strukturen in den letzten Jahren, gibt es derzeit insgesamt mehr als 225 000 Einträge in der Proteindatenbank (Abbildung 1).
|
|
| Abbildung 1. Die Proteindatenbank. Anwachsen der experimentell bestimmten 3D-Strukturen von Makromolekülen. https://www.rcsb.org/stats/growth/growth-released-structures. (Grafik heruntergeladen am 12.10.2024). |
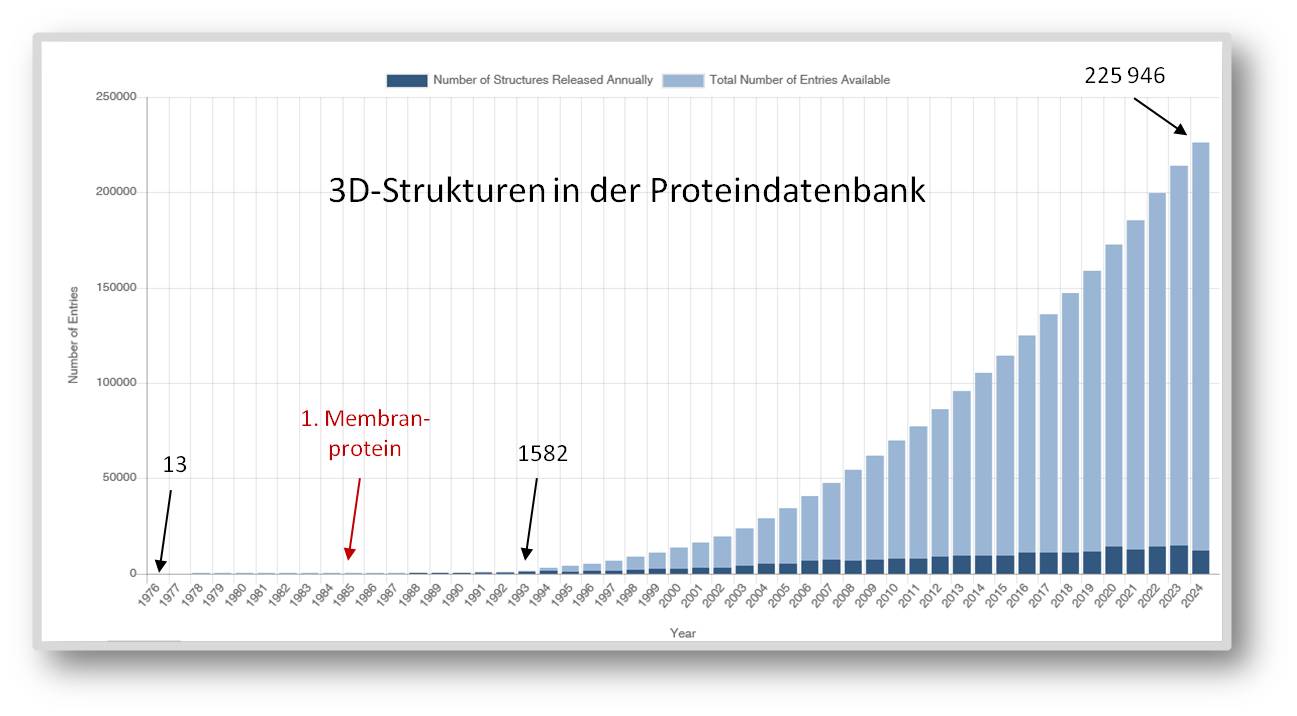
Über 200 000 Proteinstrukturen von diversen Lebensformen - das klingt nach viel, ist aber eine geringe Zahl verglichen mit den bereits mehr als 200 Millionen Proteinsequenzen, die - mit rasant verbesserten DNA-Sequenzierungstechniken - bisher in diversen Organismen identifiziert wurden. Die experimentelle Strukturaufklärung hinkt nach, ist ja noch immer recht mühsam und für einige Proteintypen (noch) kaum möglich.
Von Anfang an stellten sich die Forscher daher die Frage, ob und wie man auf Basis der Aminosäuresequenzen Vorhersagen für die Strukturen treffen könne.
..... und eine Hierarchie der Strukturen
Die Abfolge der 20 unterschiedlichen Aminosäuren in den linearen Ketten - der Primärstruktur- ist in der DNA kodiert. Dass solche Ketten dazu tendieren regelmäßige, durch Wasserstoffbrücken stabilisierte Substrukturen, alpha-Helices und beta-Faltblatt-Elemente, zu bilden, wurde von amerikanischen Chemiker Linus Pauling 1951 postuliert und deren Vorliegen in den ersten aufgeklärten Proteinstrukturen bestätigt (Pauling erhielt 1954 den Nobelpreis für Chemie). Die sogenannte Sekundärstruktur umfasst diese Substrukturen und daneben ungeordnete Bereiche. Wie sich Sekundärstrukturen dann falten, wird durch Lage und chemische Eigenschaften der einzelnen Atome, funktionellen Gruppen und Substrukturen bestimmt, in anderen Worten: wie sich diese anziehen oder abstoßen. Aus der Sekundärstruktur entsteht die für jedes Protein spezifische dreidimensionale Tertiärstruktur, die sogenannte native Konformation, welche für die Funktion bestimmend ist. Wechselwirkungen zwischen gleichen und anderen Arten von Proteinen führen schließlich zu Quartärstrukturen. Abbildung 2.
|
|
| Abbildung 2. Proteinfaltung dargestellt im Bändermodell. In der Sekundärstruktur haben sich aus der Aminosäurenkette die Substrukturen alpha-Helix und beta-Faltblatt (Pfeile in Richtung N zu C-Terminus) und ungeordnete Elemente gebildet. Die aus den Elementen der Sekundärstruktur zusammengesetzte Tertiärstruktur - die native Konformation - bestimmt die Funktion des Proteins. Zusammenlagerung von mehreren (unterschiedlichen) Proteinen durch Wechselwirkungen zwischen den Proteinen erzeugt Quartärstrukturen, Beispiel: Hämoglobin. (Quelle: aus gemeinfreien Bildern zusammengesetzt.) |
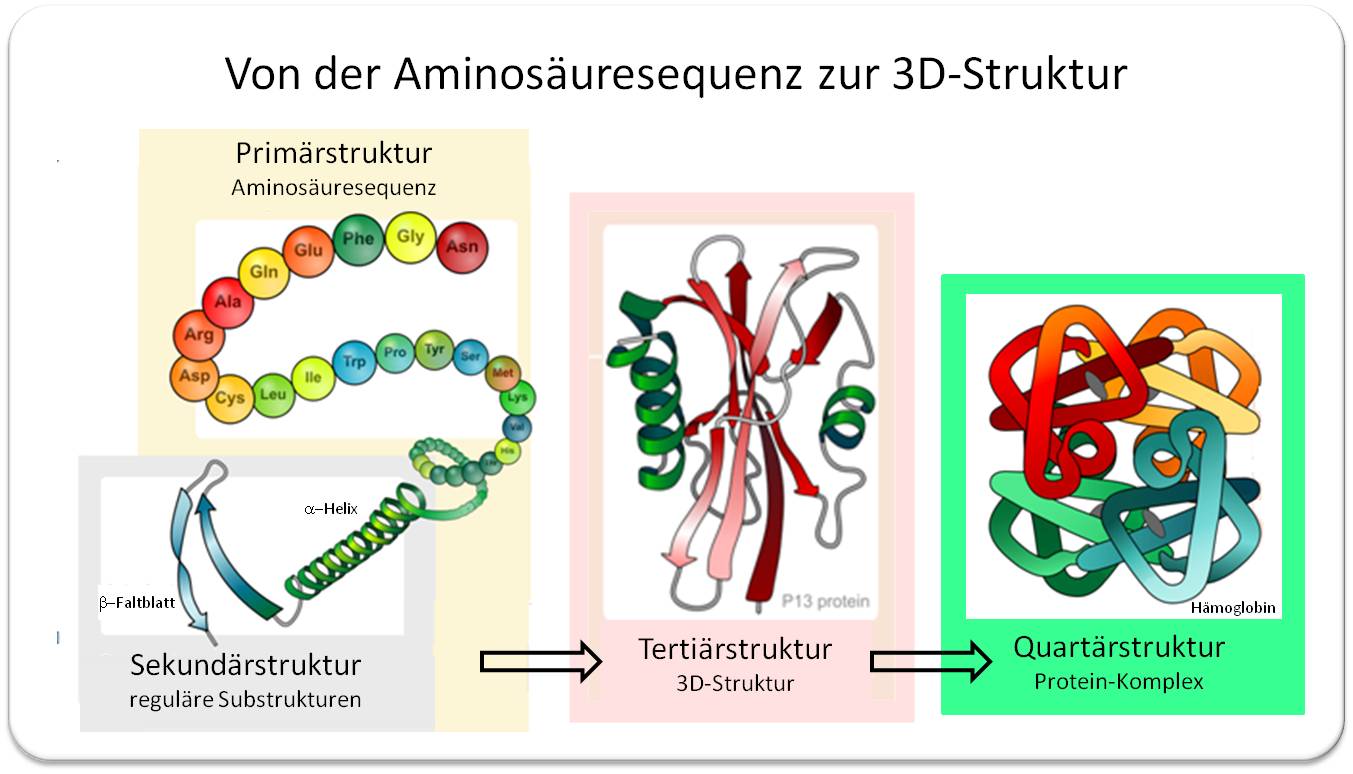
Um die räumlichen Voraussetzungen für biochemische Funktionen - beispielsweise die Bindung eines kleinen Moleküls - zu schaffen, ist eine minimale Kettenlänge von 40 - 50 Aminosäuren erforderlich.
Zur Vorhersage der 3D-Struktur
Eine Vorhersage der Tertiärstruktur auf der Basis der potentiellen Wechselwirkungen der einzelnen Atome würde selbst bei einem kleinen Protein mit 100 Aminosäuren eine praktisch unbegrenzte Anzahl möglicher Strukturen schaffen und alle Zeit würde nicht reichen, um diese bis zur energetisch günstigsten, nativen Konformation durchzuspielen. Dass Proteine nicht über ein solches Ausprobieren ihre native Konformation finden, ist offensichtlich: unter physiologischen Bedingungen in Zellen dauert es Millisekunden von der neu synthetisierten Aminosäurenkette bis zur funktionellen 3D-Struktur.
Der amerikanische Biochemiker Christian Anfinsen hatte sich seit den 1950er-Jahren mit dem Zusammenhang zwischen Primärsequenz und 3D-Struktur von Proteinen beschäftigt und herausgefunden, dass die Faltung ein vorbestimmter Prozess ist: Als er 1961 das Enzym Ribonuklease unter verschiedensten ausgeklügelten Bedingungen reversibel denaturierte, d.i. die dreidimensionale Form in eine offene, enzymatisch inaktive Kette überführte und diese dann wieder sich falten ließ, entdeckte er, dass das entstandene Produkt jedes Mal die ursprüngliche Gestalt wieder angenommen hatte und enzymatisch aktiv war. Anfinsen schloss daraus, dass die native Konformation eines Proteins ausschließlich durch seine Aminosäuresequenz bestimmt wird, die wiederum im betreffenden Gen kodiert ist.
Anfinsens Hypothese fand weite Bestätigung durch andere Labors und wurde zum Startschuss weltweiter Bemühungen aus der Kenntnis der Aminosäuresequenzen die 3D-Strukturen von Proteinen vorherzusagen. Damit würde man ja nicht nur die mühsame, langdauernde Röntgenkristallographie umgehen, sondern rasch Aussagen für alle bekannten Proteine treffen können, auch für solche, bei denen experimentelle Verfahren (noch) nicht anwendbar sind. Die Bedeutung solcher Vorhersagen für verschiedenste Gebiete der Lebenswissenschaften in akademischer Forschung und Industrie war evident.
Zur rascheren Entwicklung von Voraussagetechniken wurde 1994 ein Projekt Critical Assessment of Protein Structure Prediction (CASP) gestartet - ein alle zwei Jahre stattfindender Wettbewerb, bei dem Forscher aus aller Welt ihre Methoden an eben aufgeklärten, noch geheim gehaltenen Strukturen ausprobierten. Beginnend bei etwa 10 % war die Übereinstimmung zwischen theoretischen Modellen und experimentell bestimmter Struktur bis 2016 noch zu niedrig (30 bis maximal 40 %), stieg aber 2018 auf fast 60 % und 2020 schließlich auf 90 %, eine Genauigkeit, die im Bereich der Genauigkeit von reproduzierten experimentelle Analysen liegt. Den sensationellen Durchbruch haben Demis Hassabis und John Jumper mit den KI-gestützten Programmen AlphaFold (2018) und AlphaFold2 (2020) erzielt, in denen sie Deep-Learning-Methoden unter Verwendung von "faltenden neuronalen Netzwerken" anwandten.
Von der Sequenz zur Struktur - der Durchbruch mit Alphafold
Demis Hassabis ist kein Unbekannter. Der britische Neurowissenschafter, Programmierer, Entwickler von Computerspielen und Schachmeister ist Mitbegründer und CEO des Startups DeepMind, das auf die Programmierung von künstlicher Intelligenz spezialisiert ist und - 2014 von Google aufgekauft- zu Google DeepMind wurde. Bekannt wurde DeepMind unter anderem durch die Programme AlphaGo und AlphaGo-Zero, welche die Weltmeister im Go-Spielen schlagen konnten [6].
2018 kam das Computerprogramm AlphaFold heraus, basierend auf der Grundlage eines neuronalen Faltungsnetzwerks und auf den Strukturen der Protein Data Bank trainiert, um eine sogenannte Distanz-Karte der Abstände zwischen den Aminosäuregruppen in der räumlichen Struktur zu erstellen. Wie erwähnt gewann AlphaFold den CASP-Wettbewerb 2018. Das Programm erhielt eine wesentliche Verbesserung als der junge Biochemiker John Jumper in DeepMind eintrat und seine Kenntnisse in Protein-Chemie und Modellierung einbrachte. Das nun von Hassabis und Jumper 2020 präsentierte Programm AlphaFold2 war auf allen damals bekannten Proteinstrukturen (Abbildung 2) und den nahezu 200 Millionen Proteinsequenzen trainiert und erreichte eine Genauigkeit von etwa 90 %, die als gleichwertig mit der experimentell erreichten angesehen wird.
Eine vereinfachte Beschreibung wie AlphaFold2 arbeitet ist in Abbildung 3 dargestellt.
|
|
| Abbildung 3. AlphaFold2: In 4 Schritten von der Aminosäuresequenz zur Proteinstruktur.( © Illustration: Johan Jarnestad/The Royal Swedish Academy of Sciences; Übersetzung des Texts: I. Schuster) |
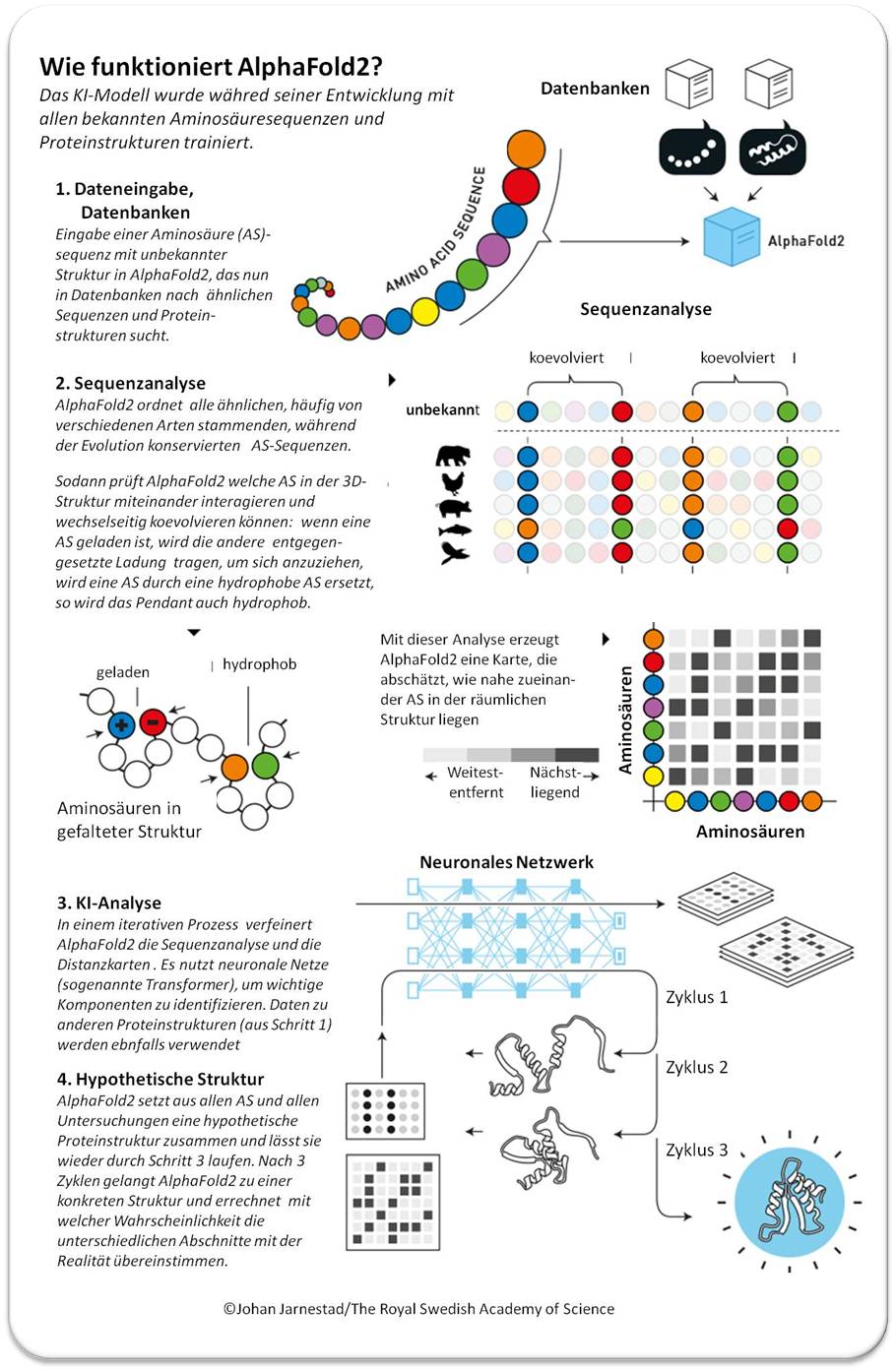
2020 wurde erstmals davon gesprochen, dass mit AlphaFold2 das Problem der Vorhersage von Proteinstrukturen aus der Primärsequenz als prinzipiell gelöst betrachtet werden könne.
Hassabis und Jumper haben mit AlphaFold2 zunächst die Struktur aller menschlichen Proteine vorhergesagt, bis 2022 rund 1 Million weiterer Strukturen - diese sind in der die Proteindatenbank PDB hinterlegt - und schließlich die Strukturen von praktisch allen 200 Millionen bislang identifizierten Proteinen. Alle diese Strukturen sind in der von Google DeepMind und dem EMBL’s European Bioinformatics Institute geschaffenen AlphaFold Datenbank öffentlich zugänglich (https://alphafold.ebi.ac.uk/))
Seit Sommer 2021 ist die Software von AlphaFold2 freigegeben (Open-Source-Lizenz). Jeder, der möchte kann nun Proteine falten. Zuvor hatte es oft Jahre gedauert, um eine Proteinstruktur zu erhalten, jetzt sind es nur noch ein paar Minuten Rechenzeit.
Mehr als 2 Millionen Forscher aus 190 Ländern haben von dem Programm schon Gebrauch gemacht. Die Tragweite dieser Möglichkeiten für Grundlagenforschung und diverse Anwendungen ist nicht absehbar.
Von der Struktur zur Sequenz - das Rosetta-Programm
Früher als Hassabis und Jumper hat sich der US-amerikanische Biochemiker David Baker, Direktor des Instituts für Protein Design an der Universität Washington, mit der Strukturvorhersage von Proteinen befasst und schon 1998 an den CASP-Wettbewerben teilgenommen. Er hatte das Computerprogramm Rosetta entwickelt, das - wie die Programme der Konkurrenten - aus den Aminosäuresequenzen die Proteinstrukturen vorhersagen sollte, diesen aber überlegen war. Baker und sein Team änderten aber bald die Strategie und gingen den umgekehrten Weg: anstatt vorherzusagen zu welcher Tertiärstruktur sich eine Primärstruktur falten würde, wollte man eine völlig neue Struktur schaffen und dann herausfinden, welche Sequenz sich zu dieser falten würde. In anderen Worten: Man wollte Rosetta so weiterentwickeln, dass man für diverse Funktionen - beispielsweise für spezielle Enzymaktivitäten - maßgeschneiderte Proteine würde designen können.
Die Machbarkeit dieses Ansatzes konnte Baker 2003 bestätigen: Er und sein Team hatten mit Top 7 ein völlig neues, in der Natur nicht vorkommendes Protein kreiert, das mit seinen alpha-Helix/ beta-Faltblatt Strukturen besonders stabil war und mit 93 Aminosäuren größer war, als alle bis dahin synthetisch hergestellten Proteine. Um den Erfolg der Software zu prüfenen, hatte man das Gen für die vorgeschlagene Aminosäuresequenz in Bakterien eingeführt und das von diesen produzierte Protein mit Hilfe der Röntgenkristallographie analysiert.
|
|
| Abbildung 4. Proteine, die mit dem Rosetta-Programm von Baker entwickelt wurden: (Bild: ©Terezia Kovalova/The Royal Swedish Academy of Sciences, Text übersetzt von I. Schuster) |

In weiterer Folge wurde durch Übernahme einer AlphaFold2 ähnlichen Deep Learning Architektur aus Rosetta das RoseTTAFold Programm, dessen Software frei zugänglich ist (https://github.com/RosettaCommons/RoseTTAFold ). Laut Homepage des Baker Instituts ist RoseTTAFold "ein "dreigleisiges" neuronales Netz, d. h. es berücksichtigt gleichzeitig Muster in Proteinsequenzen, die Art und Weise, wie die Aminosäuren eines Proteins miteinander interagieren, und die mögliche dreidimensionale Struktur eines Proteins. In dieser Architektur fließen ein-, zwei- und dreidimensionale Informationen hin und her, so dass das Netzwerk gemeinsam Schlussfolgerungen über die Beziehung zwischen den chemischen Bestandteilen eines Proteins und seiner gefalteten Struktur ziehen kann." (https://www.ipd.uw.edu/2021/07/rosettafold-accurate-protein-structure-prediction-accessible-to-all/)
Seitdem wurden mit RoseTTAFold Tausende neue Proteine designt, Strukturen, die für diversesten Anwendungen in Industrie, Umwelt und Gesundheit relevant sind. Einige dieser spektakulären Strukturen sind in Abbildung 4 dargestellt.
Ausblick
Das Nobel-Kommittee für Chemie schreibt [2]:
„Die Errungenschaften von David Baker, Demis Hassabis und John Jumper auf dem Feld des computergestützten Proteindesigns und der Proteinstruktur-Entschlüsselung sind fundamental. Ihre Arbeit hat eine neue Ära der biochemischen und biologischen Forschung eröffnet, in der wir nun Proteinstrukturen auf eine Weise entwerfen und vorhersagen können, wie es nie zuvor möglich war. Damit wurde ein lang gehegtes Ziel endlich erreicht, und die Auswirkungen werden weitreichende Konsequenzen haben.“
[1] The Nobel Prize in Chemistry 2024, Popular Science Background: They have revealed proteins’ secrets through computing and artificial intelligence.https://www.nobelprize.org/uploads/2024/10/popular-chemistryprize2024-3.pdf
[2] The Nobel Prize in Chemistry 2024, Scientific Background: Computational Protein Design and Protein Structure Prediction. https://www.nobelprize.org/uploads/2024/10/advanced-chemistryprize2024.pdf
[3] Peter Schuster, 04.03.2016: Die großen Übergänge in der Evolution von Organismen und Technologien
[4] Bernhard Rupp, 04.04.2014: Wunderwelt der Kristalle — Von der Proteinstruktur zum Design neuer Therapeutika
[5] PDP-Protein Data Bank: History:https://www.rcsb.org/pages/about-us/history
[6] Norbert Bischofberger, 16.08.2018: Mit Künstlicher Intelligenz zu einer proaktiven Medizin
- Printer-friendly version
- Log in to post comments