Do. 25.09.2025 — Ricki Lewis

![]()
Es kommt selten vor, dass Ergebnisse klinischer Studien so aufregend sind wie die, die das niederländisch-amerikanische Biotech-Unternehmen UniQure eben bekannt gegeben hat. [1] Erste Ergebnisse nach einer dreijährigen klinischen Studie in den USA, Großbritannien und Europa deuten darauf hin, dass die derzeit unter dem Namen AMT-130 bekannte Behandlung das Fortschreiten der Huntington-Krankheit (HK) deutlich verlangsamt. Laut einer Pressemitteilung von UniQure könnte sie bereits 2026 in den USA zugelassen werden.*
Eine Variation zum Thema Gentherapie: Mikro-RNAs
Die Strategie nutzt die Mikro-RNA-Technologie, um winzige RNA-Stücke auf die mRNA-Moleküle zu stempeln, welche die Informationen in einem zu langen, die Huntington-Krankheit auslösenden Gen tragen, das für ein Protein namens Huntingtin kodiert (s.u,).
Das übergroße Protein verstopft Teile des Gehirns, die für die Bewegung entscheidend sind.
Die neue Behandlung verlangsamt den Verfall, sodass das, was normalerweise innerhalb eines Jahres geschieht, nun vier Jahre dauert. Das ist eine Veränderung, die das Leben der Familien der rund 75.000 Menschen mit Huntington-Krankheit in den USA, Großbritannien und Europa sowie vieler anderer Menschen auf der ganzen Welt und ihrer Familien grundlegend verändern würde.
Ich schreibe seit Jahrzehnten über Huntington [2], und dies ist meiner Meinung nach der erste echte Hoffnungsschimmer für Familien, die von dieser verheerenden Krankheit betroffen sind. Bereits 2021 habe ich hier bei DNA Science über die UniQure-Forschung berichtet, die zu der aktuellen klinischen Studie geführt hat, und vor zwei Wochen habe ich über einen Vater geschrieben, der seine aussergewöhnliche, an Huntington gestorbene Tochter würdigt.
AMT-130 ist eine Art der Gentherapie, die in einer ein Mal Anwendung in das Gehirn injiziert wird.
Die Biologie der Huntington-Krankheit
Die Huntington-Krankheit ist eine von mehr als vierzig „Expanding-Repeat” (expandierende Wiederholungen, Anm. Redn,) -Erkrankungen, bei denen ein Gen von Generation zu Generation expandiert - länger wird. Ab einer bestimmten Anzahl von Wiederholungen in der DNA-Sequenz verstopft das davon kodierte Protein Teile des Gehirns, die unter anderem die Motorik steuern.
Das Gen, das bei Expansion die Huntington-Krankheit verursacht, heißt Htt und kodiert für das Protein Huntingtin. Das Gen enthält normalerweise am Anfang seiner Sequenz bis zu 35 Kopien eines DNA-Tripletts (GTC, das für eine mRNA mit CAG codiert), das der Zelle sagt, dass sie die Aminosäure Glutamin in die wachsende Proteinkette einfügen soll. (In den meisten Artikeln ist von CAG-Wiederholungen in der DNA die Rede, aber das ist eine zu starke Vereinfachung, denn tatsächlich befindet sich das CAG in der mRNA, die von der DNA codiert wird.)
|
|
|
Die Huntington-Krankheit beruht auf einer Triplett-Wiederholung in der DNA, die zu einer zu einer zu häufigen Abfolge der für die Aminosäure Glutamin kodierenden Nukleotide Cytosin-Adenin-Guanin (CAG) in der mRNA transkribiert wird. Ein zu langes Gen kodiert für ein Protein, welches das Striatum des Gehirns verstopft. |
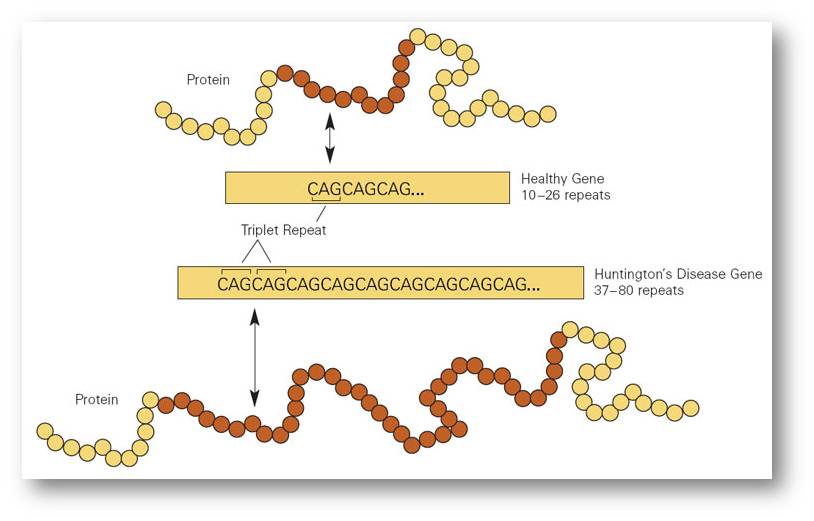
Die zusätzlichen Glutamine im Huntingtin-Protein lösen eine Kaskade von Zerstörungen aus, die nach und nach die „mittelgroßen stacheligen Neuronen“ in den Motorikzentren des Gehirns zerstören. Das expandierte Protein kann sich nicht richtig falten und haftet an sich selbst und anderen Proteinen. Es blockiert die Axone in den Neuronen des Striatums des Gehirns und verhindert so die Verteilung wichtiger Wachstumsfaktoren. Die weiße Substanz des Gehirns schrumpft.
Das Ergebnis sind die unkontrollierbaren Bewegungen der Huntington-Krankheit, die zunächst subtil sind, dann immer ausladender und tanzähnlicher werden und schließlich in eine unheimliche Reglosigkeit übergehen. Auch psychische Symptome treten auf, wie Wut, Unruhe, Frustration und Verwirrung.
Menschen können viele Jahre mit Huntington leben. Die Krankheit hat verheerende Auswirkungen auf die gesamte Familie, da autosomal-dominante Mutationen sowohl Frauen als auch Männer betreffen, nur von einem Elternteil vererbt werden müssen und die Übertragung nur dann unterbrochen wird, wenn zufällig das funktionsfähige Gen und nicht das mutierte Gen weitergegeben wird. 2013 habe ich eine Familie mit der „grausamen Mutation” der juvenilen Form von Huntington beschrieben – sie traf einen Vater und seine drei jungen Töchter [2].
Nutzung und Steuerung einer Mikro-RNA
Die neue Behandlung ist eine Variante der Gentherapie. Dabei werden viele Kopien einer kurzen RNA-Sequenz, einer sogenannten Mikro-RNA, in einer 12- bis 14-stündigen Operation über einen Virus-Vektor in das Gehirn eingebracht. Die Mikro-RNAs binden sich dann an die Messenger-RNA-Moleküle (mRNA), die aus dem Huntingtin-Gen transkribiert werden. Das funktioniert ein wenig wie molekularer Reissverschluss.
Die Behandlung wurde zunächst in präklinischen Studien an Mäusen und Schweinen getestet. Sehr wichtig für die Arzneimittelentwicklung sind auch natürliche Studien, in denen über Jahrzehnte hinweg Informationen zum Krankheitsverlauf bei vielen Menschen mithilfe von Biomarkern, Scans und Bewertungsskalen gesammelt werden. Bei Huntington können Familienmitglieder seit 1993 einen „Prämanifestationstest” machen, um herauszufinden, ob sie die Mutation geerbt haben, bevor Symptome auftreten. Das ist eine schwere Entscheidung. Aber diese selbstlosen und mutigen Menschen sind für die Planung und Durchführung klinischer Studien von entscheidender Bedeutung.
In der klinischen Studie zu AMT130 wurden eine niedrige Dosis, eine hohe Dosis und Scheinbehandlungen bei Patienten im Frühstadium der Erkrankung untersucht.
In den USA erhielten 6 Teilnehmer eine niedrige Dosis, 10 eine hohe Dosis und 10 eine Scheinbehandlung (eine kurze Injektion, um die Behandlung vorzutäuschen). In Europa erhielten 7 Patienten die hohe Dosis und 6 die niedrige Dosis. Weitere Personen nehmen noch an der Studie teil. Es überrascht nicht, dass der Wert der UniQure-Aktie in die Höhe schoss.
Anzeichen für Erfolg
Bei Patienten, die die neue Behandlung erhielten, verlangsamte sich im Verlauf von 3 Jahren das Fortschreiten der Krankheit. Die leitende Forscherin Sarah Tabrizi, Professorin für klinische Neurologie und Direktorin des Huntington's Disease Center am University College London, bezeichnet die Ergebnisse als „spektakulär” und alle Erwartungen übertreffend.
Die Teilnehmer der Gruppe mit der höheren Dosis zeigten nach Ablauf der drei Jahre eine Verlangsamung des Krankheitsverlaufs um 75 Prozent, gemessen anhand einer Bewertungsskala namens cUHDRS. Dies war der primäre Endpunkt der Studie; selbst eine geringere Verlangsamung wäre bereits eine großartige Nachricht für diese Erkrankung gewesen, deren Bekämpfung seit vielen Jahrzehnten erfolglos war.
Eine anderes Maß, eine Skala der Gesamtfunktionsfähigkeit (TFC), zeigte eine Verlangsamung des Krankheitsverlaufs um 60 Prozent. Der TFC-Wert, der von 0 (vollständige Arbeitsunfähigkeit) bis 13 (normal) reicht, berücksichtigt Fähigkeiten in den Bereichen Beschäftigung oder Beruf, Umgang mit finanziellen Angelegenheiten, Erledigung von Hausarbeiten, Aktivitäten des täglichen Lebens und erforderlicher Pflegebedarf.
Ein weiteres Bewertungstool ist ein Biomarker für Neurodegeneration in der Rückenmarksflüssigkeit, das sogenannte Neurofilament-Leichtprotein. Umso schwerer die Erkrankung, desto höher der NfL-Wert. Die Werte sanken im Durchschnitt um 8,2 Prozent.
Ein weiteres Plus: die Behandlung erwioes sich als sicher und verträglich. Dies ist das erste Licht am Ende eines langen, dunklen Tunnels.
Dr. Tabrizi sagt: „Ich glaube, dass diese bahnbrechenden Daten die bislang überzeugendsten in diesem Bereich sind und mögliche krankheitsmodifizierende Wirkungen bei der Huntington-Krankheit hervorheben, die dringend notwendig sind. AMT-130 hat das Potenzial, das Fortschreiten der Krankheit deutlich zu verlangsamen – es bietet damit den von dieser verheerenden Krankheit betroffenen Menschen und Familien die lang ersehnte Hoffnung.“
Natürlich wird der Preis hoch sein, aber die Möglichkeit, diesen neurologischen Angriff, der die Auswirkungen von Parkinson, ALS und Alzheimer vereint, zu verlangsamen und vielleicht eines Tages zu stoppen, ist unbezahlbar.
Wenn alles gut geht, könnten Familien mit Huntington in einem Jahr zumindest in der Lage sein, den Verlauf dieser schrecklichen Krankheit zu verlangsamen.
*Der Artikel ist erstmals am 25.September 2025 in PLOS Blogs - DNA Science Blog unter dem Titel "Finally, a One-Time Treatment that May Slow the Course of Huntington’s Disease" erschienen (https://dnascience.plos.org/2025/09/25/finally-a-one-time-treatment-that-may-slow-the-course-of-huntingtons-disease/ und steht unter einer cc-by Lizenz . Die Autorin hat sich freundlicherweise mit der Übersetzung ihrer Artikel durch ScienceBlog.at einverstanden erklärt. Die Übersetzung folgt so genau als möglich der englischen Fassung.
[1] UniQure Press Release, 24.09.2025: uniQure Announces Positive Topline Results from Pivotal Phase I/II Study of AMT-130 in Patients with Huntington’s Disease. https://www.uniqure.com/investors-media/press-releases
UniQure: AMT-130 Administration. Video, 1:22 min. https://www.youtube.com/watch?v=qILELaiWtcE
[2] Ricki Lewis über Huntington Krankheit: Links zu > 11 Artikel in https://dnascience.plos.org/?search=huntington
- Printer-friendly version
- Log in to post comments