Fr, 24.05.2013 - 11:13 — Peter Schuster
![]()
 Die Vermehrung von Viren ist durch eine sehr hohe Mutationsrate geprägt. Dabei entstehen genetisch uneinheitliche Populationen , sogenannte Quasispezies, die sich in einem dynamischen Gleichgewicht von Mutation und Selektion befinde und damit einem Evolutionsprozeß unterliegen, der u.a. erhöhte Infektiosität und Pathogenität mit sich bringt. Eine weitere Erhöhung der Mutationsrate durch geeignete mutagene Verbindungen kann jedoch zur Auslöschung der Quasispezies-Populationen führen. Letale Mutagenese erscheint daher erfolgversprechend als eine neue Strategie im Kampf gegen virale Infektionen und deren Ausbreitung.
Die Vermehrung von Viren ist durch eine sehr hohe Mutationsrate geprägt. Dabei entstehen genetisch uneinheitliche Populationen , sogenannte Quasispezies, die sich in einem dynamischen Gleichgewicht von Mutation und Selektion befinde und damit einem Evolutionsprozeß unterliegen, der u.a. erhöhte Infektiosität und Pathogenität mit sich bringt. Eine weitere Erhöhung der Mutationsrate durch geeignete mutagene Verbindungen kann jedoch zur Auslöschung der Quasispezies-Populationen führen. Letale Mutagenese erscheint daher erfolgversprechend als eine neue Strategie im Kampf gegen virale Infektionen und deren Ausbreitung.
Basierend auf fulminanten Erfolgen in der Bekämpfung von Infektionskrankheiten ging man in der Mitte des vorigen Jahrhunderts davon aus, daß die Erreger dieser Krankheiten wohl bald ausgerottet sein würden. Zwar wurden vereinzelt Veränderungen der Erregerstämme detektiert, diesen aber kaum Beachtung geschenkt. Erst der systematische Einsatz molekularbiologischer Methoden, vor allem die genetische Analyse der Mikroorganismen, zeigte wie schnell und in welchem Ausmaß Veränderungen eintreten, die vormals effektive antibakterielle und antivirale Strategien unwirksam werden lassen.
Wie Viren sich vermehren
Viroide (s.u.) und Viren können als non-plus-ultra Parasiten angesehen werden, welche – allein nicht lebensfähig - den Wirt ausnützen indem sie in seine Zellen eindringen und deren Stoffwechsel für ihre eigene Vermehrung umfunktionieren. Grundlegend für diese Vermehrung ist die erfolgreiche Kopierung (Replikation) des viralen Genoms, welches aus Ribonukleinsäure (RNA)- oder Desoxyribonuleinsäure (DNA)-Molekülen besteht (Abbildung 1). Erfolgreich bedeutet dabei, daß die viralen Nachkommen ebenfalls fähig sind sich zu vermehren.
 |
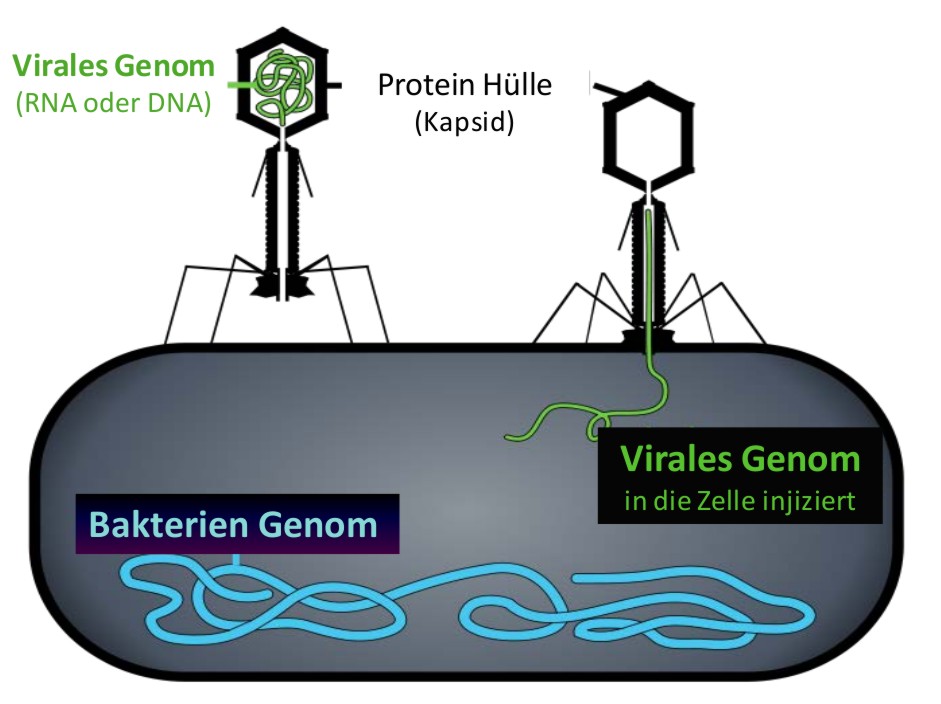 |
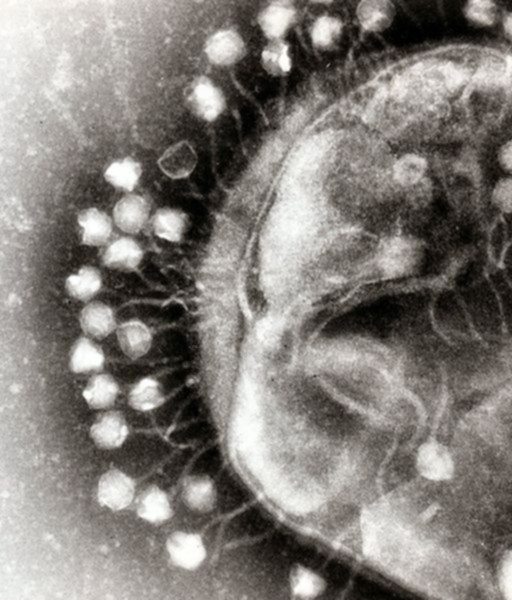
| Abbildung 1. Bakteriophage heftet sich an die Zellwand eines Bakteriums an (links, elektronenmikroskopisches Bild) und injiziert sein Genom in die Wirtszelle (rechts). (Bilder modifiziert nach Wikimedia Commons) | |
Die Vermehrung von Viren ist ein komplexer Vorgang, da bereits das Kopieren der Nukleinsäuren ein aus vielen Einzelschritten bestehender Prozeß ist, wobei der Stoffwechsel der Wirtszelle mehr oder weniger in Anspruch genommen wird (Abbildung 2). Viroide – Pflanzenpathogene -, die überhaupt nur aus dem „nackten“ RNA-Molekül bestehen, sind für ihre Replikation vollständig auf die biochemische Ausstattung und die Reaktionsmechanismen des Wirts angewiesen. 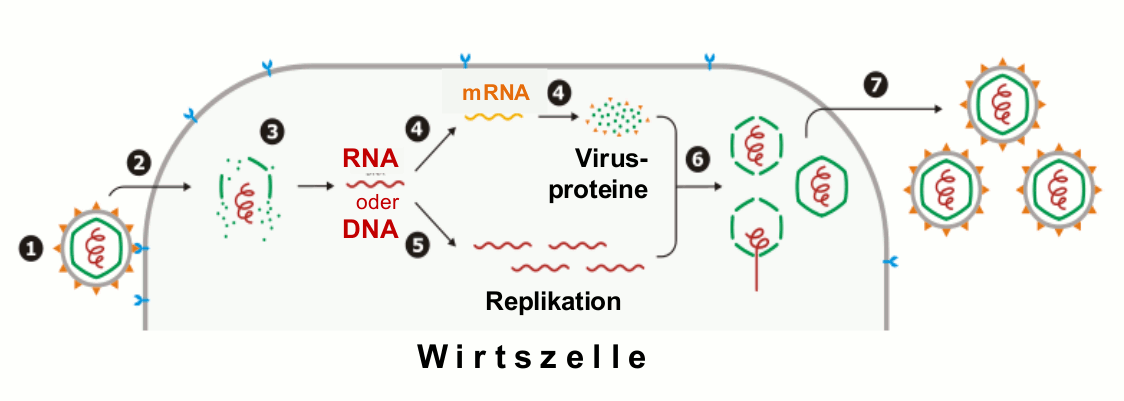 Abbildung 2. Replikationszyklus eines Virus. 1: Anheften an die Wirtszelle, 2: Aufnahme in die Zelle und 3: Abstreifen der Umhüllung (uncoating). 4: Transkription des Genoms und Umsetzung der mRNA zu viralen Proteinen (Translation), 5: Vervielfältiung (Replikation) des viralen Erbmaterials (RNA oder DNA), 6: Aggregation von Hüllproteinen – Bildung neuer Partikel, 7: Freisetzung (stark vereinfachte Darstellung, modifiziert nach commons, wikimedia).
Abbildung 2. Replikationszyklus eines Virus. 1: Anheften an die Wirtszelle, 2: Aufnahme in die Zelle und 3: Abstreifen der Umhüllung (uncoating). 4: Transkription des Genoms und Umsetzung der mRNA zu viralen Proteinen (Translation), 5: Vervielfältiung (Replikation) des viralen Erbmaterials (RNA oder DNA), 6: Aggregation von Hüllproteinen – Bildung neuer Partikel, 7: Freisetzung (stark vereinfachte Darstellung, modifiziert nach commons, wikimedia).
Das Genom von RNA-Viren ist besonders klein, es enthält bloß zwischen 3000 und 33000 Nukleotide. Derart einfache Genome kodieren nur für einige wenige Proteine: üblicherweise sind das i) ein Enzym, das spezifisch die virale RNA repliziert (RNA-Replicase), ii) ein zweites Protein, welches eine Hülle um die virale RNA bildet und iii) ein weiteres Protein, welches die Auflösung - Lyse - der Wirtszelle und damit die Freisetzung des Virus einleitet. Bei Bakteriophagen, das sind Viren, die sich in Bakterienzellen vermehren, weist die genomische RNA Ähnlichkeit mit den Messenger-RNAs der Wirtszellen auf. Sie wird deshalb von der Proteinsynthese-Maschinerie der Wirtszelle - den Ribosomen - erkannt und unmittelbar nach Eintritt in die Wirtszelle zu Virusproteinen umgesetzt (translatiert).
Bestimmend für den Vermehrungszyklus von Viren: Mutation und Selektion
Das System Bakteriophage – Bakterienzelle stellt ein einfaches Modell dar, mit welchem die Mechanismen der Viren-Vermehrung untersucht werden können. An diesem Modell (Qβ-Phage – Escherichia coli Bakterie) hat Charles Weissmann bereits in den 1970er-Jahren gezeigt, daß der Vermehrungszyklus bestimmt wird durch die Struktur der Virus-RNA und die Dynamik, mit der sich diese faltet und entfaltet.
Generell erfolgt der Kopiervorgang eines Genoms nicht fehlerfrei. Unter allen Spezies zeigen Viroide und Viren die höchsten Mutationsraten (Abbildung 3). Diese beträgt etwas weniger als eine Mutation pro Genom und Replikation.
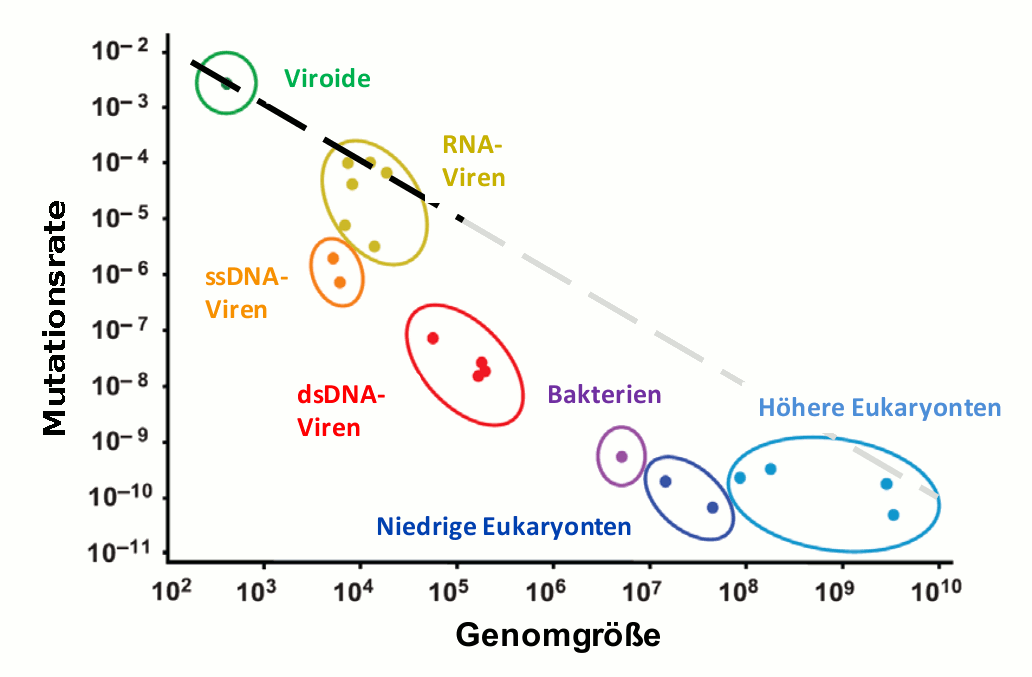 Abbildung 3. Die Genauigkeit mit der ein Genom kopiert wird (Mutationsrate), hängt von der Größe des Genoms (Anzahl der Nukleotide) ab. Mutationsrate: Anzahl der Veränderungen/Nukleotid. ssDNA-Viren: Einfachstrang-DNA-Viren , dsDNA -Viren: Doppelstrang-DNA-Viren , Bakterien: E.coli, niedrige Eukaryonten: Hefen, höhere Eukaryonten: Caenorhabditis elegans, Drosophila melanogaster, Maus, und Mensch. Ein Viroid kann aus nur 250 Nukleotiden bestehen, das humane Genom ist 3,3 Milliarden Nukleotidpaare lang, (Quelle: S. Gago et al. Science, 2009, 323 (5919) 1308; powerpoint slide for teaching)
Abbildung 3. Die Genauigkeit mit der ein Genom kopiert wird (Mutationsrate), hängt von der Größe des Genoms (Anzahl der Nukleotide) ab. Mutationsrate: Anzahl der Veränderungen/Nukleotid. ssDNA-Viren: Einfachstrang-DNA-Viren , dsDNA -Viren: Doppelstrang-DNA-Viren , Bakterien: E.coli, niedrige Eukaryonten: Hefen, höhere Eukaryonten: Caenorhabditis elegans, Drosophila melanogaster, Maus, und Mensch. Ein Viroid kann aus nur 250 Nukleotiden bestehen, das humane Genom ist 3,3 Milliarden Nukleotidpaare lang, (Quelle: S. Gago et al. Science, 2009, 323 (5919) 1308; powerpoint slide for teaching)
Die Genauigkeit, mit welcher die virale RNA-Replicase die Nukleinsäure kopiert, ist ausschlaggebend dafür, ob brauchbare Information erzeugt wird, die zur Reproduktion von Nachkommenschaft befähigt. Der reproduktive Erfolg einer Variante – die sogenannte Fitness - wird üblicherweise an Hand ihrer Nachkommen in der nächsten Generation bestimmt. Eine Mutation in einem Gen führt zu einer Änderung in der Sequenz der Nukleotide und daraus kann sich eine Änderung in der Aminosäuresequenz des kodierten Proteins ergeben. Derartige Änderungen können die Struktur und damit die Funktion des Proteins in einer Weise beeinflussen, welche den reproduktiven Erfolg i) kaum beeinträchtigt (neutrale Mutation), ii) schmälert oder unmöglich macht, oder iii) in vorteilhafter Weise erhöht. Die im letzteren Fall entstehende Variante hat mehr direkte Nachkommen, als die um die Ressourcen desselben Systems konkurrierenden neutralen Varianten und das vererbte günstige Merkmal führt zu mehr und mehr Nachkommen in den folgenden Generationen während ungünstige Mutationen zu einem raschen Absinken der Häufigkeit führen mit der betroffene Varianten in der Gesamtpopulation angetroffen werden. Diese natürliche Selektion steuert den Evolutionsprozeß des Virus.
Quasispezies - Ein Schwarm von Varianten
Frühe, sehr arbeitsaufwändige analytische Untersuchungen an Viruspopulationen, die sich aus einer definierten Q-Phagen-RNA auf dem Bakterienrasen (E. coli) entwickelt hatten, zeigten: die Population RNA-Moleküle ist nicht einheitlich, sondern es existiert eine große Anzahl unterschiedlicher Sequenzen, die sich um 1 – 2 Mutationen von der ursprünglichen Sequenz unterscheiden. Dieses Ergebnis war überraschend, hatte doch bis dahin jede einschlägig arbeitende Gruppe die Sequenz „ihres“ Virus als fix angesehen. Der offensichtliche Schwarm an Varianten, der nun gleichzeitig in einer Virenpopulation nachgewiesen wurde, entsprach dem Konzept der sogenannten Quasispezies, deren Existenz Manfred Eigen in seiner molekularen Theorie der Evolution etwa gleichzeitig mit der Entdeckung der Heterogenität der Viruspopulationen postuliert hatte.
Basierend auf den Prinzipien der Darwin’schen Theorie von Mutation und natürlicher Selektion berücksichtigt das Eigen’sche Konzept auch die dynamischen Eigenschaften der Replikationsprozesse und die Wechselwirkungen zwischen den Molekülen und ist direkt auf die Replikation viraler RNA anwendbar:
Bei hinreichend genauer Kopierung der RNA und genügend lange nach der Infektion stellt sich eine stationäre Quasispezies –Mutanten Verteilung in der Viruspopulation ein: die Virusvariante mit der größten Fitness – die Mastersequenz - liegt in höchster Konzentration vor, daneben existiert ein Schwarm an Varianten. Wie hoch die Anteile von Mastersequenz und Varianten in der Quasispezies sind, hängt von der Mutationsrate und den Fitnessunterschieden ab (Abbildung 4):
Exaktes Kopieren der Mastersequenz produziert eine homogene Population, welche ausschließlich aus der Mastersequenz besteht. Mit steigender Mutationsrate nimmt der Anteil der Varianten stetig zu bis ein kritischer Wert erreicht wird. An der Fehlerschwelle breiten sich die Kopierfehler in der Population so rasch aus, dass sich keine stationäre Mutantenverteilung mehr einstellen kann. Die Konsequenz beim Überschreiten der Fehlerschwelle ist ein sich Auflösen der Populationsstruktur, da alle Mutanten gleich wahrscheinlich sind und die Population zur Gleichverteilung strebt, die in der Realität niemals eintreten kann.
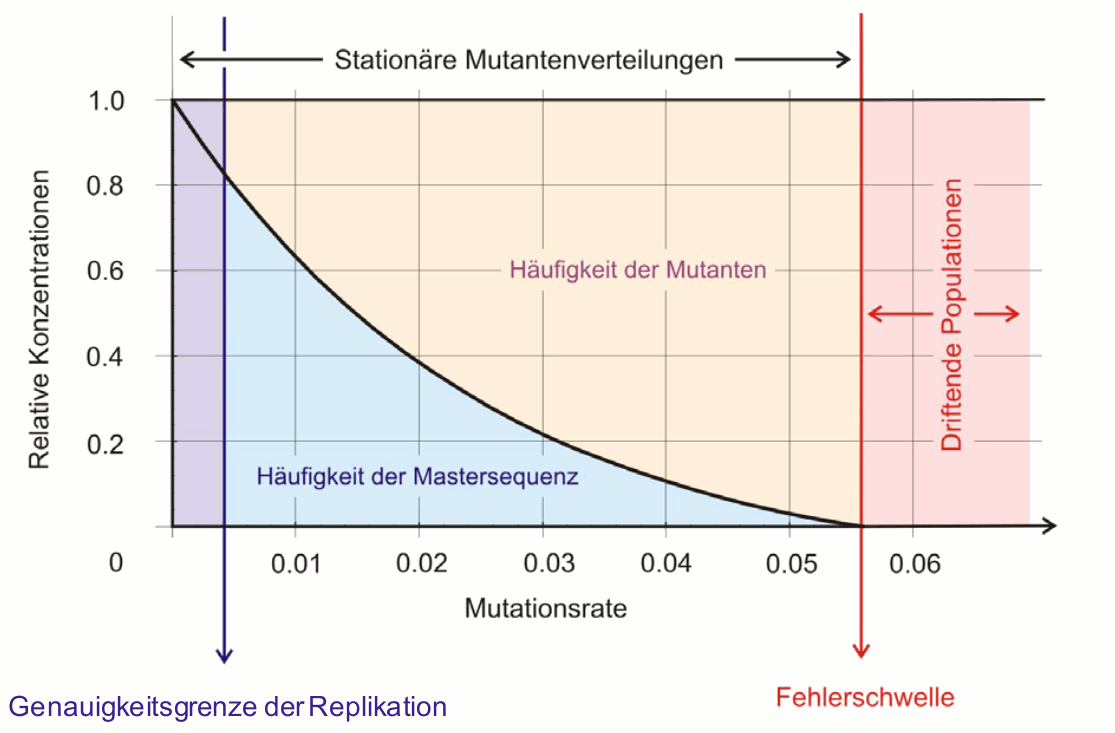 Abbildung 4. Letale Mutagenese: Ausgehend von einem exakten (in der Realität unmöglichen) Kopieren der RNA führt eine steigende Mutationsrate (Kopierfehler während der Replikation) zu einem stetigen Absinken des Anteils an Mastersequenz und einem reziproken Anwachsen des Anteils bis eine Fehlerschwelle erreicht wird, ab der alle Mutanten gleichverteilt sind und die Populationsstruktur sich auflöst („driftet“). (Die Mutationsrate ist angegeben in: Veränderungen pro Replikation je Nukleotid).
Abbildung 4. Letale Mutagenese: Ausgehend von einem exakten (in der Realität unmöglichen) Kopieren der RNA führt eine steigende Mutationsrate (Kopierfehler während der Replikation) zu einem stetigen Absinken des Anteils an Mastersequenz und einem reziproken Anwachsen des Anteils bis eine Fehlerschwelle erreicht wird, ab der alle Mutanten gleichverteilt sind und die Populationsstruktur sich auflöst („driftet“). (Die Mutationsrate ist angegeben in: Veränderungen pro Replikation je Nukleotid).
Die Alternative zum Überschreiten der Fehlerschwelle ist ein Aussterben der Population durch Akkumulation letaler, das bedeutet nicht vermehrungsfähiger Varianten. Auch diese Situation kann durch Erhöhen der Mutationsrate erreicht werden. In Abbildung 5 sind die beiden Wege zur Auslöschung der Virenpopulation einander gegenübergestellt. Je höher die Zahl der letalen Positionen in der Sequenz der viralen Nukleinsäure ist, umso eher tritt Aussterben durch akkumulierte Letalität ein.
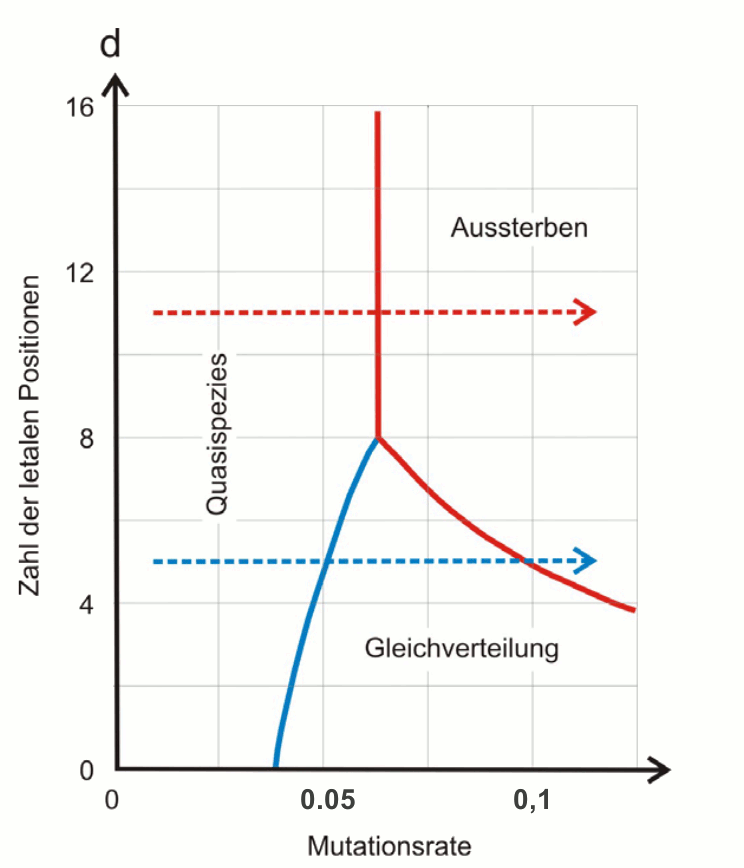
Abbildung 5. Letale Mutagenese: bei steigender Mutationsrate (Kopierfehler während der Replikation) nimmt i) der Anteil an Varianten zu, bis eine Fehlerschwelle erreicht wird, ab der alle Mutanten gleichverteilt sind und die Populationsstruktur sich auflöst (blau strichlierter Pfeil; siehe Abbildung 4) und ii) die Zahl letaler Veränderungen im Genom sich erhöht und damit die Entstehung nicht vermehrungsfähiger Varianten (rot strichlierter Pfeil). In beiden Fällen resultiert das Aussterben der Viruspoulationen. (Mutationsrate: Veränderungen pro Replikation je Nukleotid)
RNA-Viren replizieren mit geringer Genauigkeit, welche wie in Abbildung 3 gezeigt in der Nähe von einem Fehler pro Replikation des gesamten Genoms (zwischen 3000 und 33 000 Nukleotide) liegt. Eine einfache Abschätzung zeigt, dass dieser hohe Werte der Mutationsrate nur wenig unterhalb der Fehlerschwelle zu liegen kommt. Evolutionsmäßig betrachtet erscheint dieser Befund überzeugend, da Viren durch die Abwehrsysteme ihrer Wirtsorganismen – Restriktionsnukleasen, Immunabwehr, etc. – einem sehr hohen Selektionsdruck ausgesetzt sind und daher auf eine möglichst große Variationsbreite der Sequenzen in der Quasispezies angewiesen sind. In der Tat bereitet die hohe Variabilität einiger humanpathogener Viren besondere Probleme bei der Therapie, Beispiele sind das Influenza A Virus, das HIV I oder das Hepatitis C Virus.
Letale Mutagenese als eine neue antivirale Strategie Krankheiten, welche durch RNA-Viren mit hoch variablen Sequenzen hervorgerufen werden, stellen wegen der raschen Ausbildung von Resistenzen gegen vorhandene Medikamente oder Vakzinen als Folge der Quasispeziesstruktur der Populationen ein enormes medizinisches Problem dar. Um den Selektionsprozeß möglichst zu verzögern, werden heute häufig Kombinationstherapien angewandt, etwa im Fall der antiretroviralen Therapie (HAART) bei HIV-infizierten Patienten. Auch hier entstehen schlußendlich aber Viren-Formen, die gegen alle Medikamente der Kombinationstherapie resistent sind und damit zumTherapieversagen. Einen Paradigmenwechsel in der antiviralen Strategie stellt die auf der Basis der oben beschriebenen Phänomene – Überschreiten der Fehlerschwelle oder Akkumulation letaler Varianten – basierende letale Mutagenese dar. Substanzen, welche die Mutationsrate des Virus erhöhen – unter ihnen die zurzeit wirksamsten antiviralen Medikamente – führen mit oder ohne Vergrößerung der Breite der Mutantenverteilung zu einer Auslöschung der Viruspopulationen und bestätigen damit die Gültigkeit des neuen Ansatzes. Derart mutagene Substanzen führten in relevanten Infektionsmodellen und auch in klinischen Studien u.a. zur Eliminierung des Polio-Virus, des mit dem Tollwut-Virus verwandten Vesicular stomatitis Virus, des Lymphozytären Choriomeningitis Virus, des Maul-und Klauenseuche Virus und des HIV-Virus. Wie Sequenzanalysen viraler RNA zeigen, dürfte die Wirksamkeit des klinisch gegen Hepatitis C angewandten Medikaments Ribavirin zumindest zum Teil auf seine mutagene Wirkung zurückzuführen sein. Einige neue Viren-mutagene Verbindungen befinden sich zur Zeit in der klinischen Entwicklung .
Literatur
Domingo, E. (1978). Nucleotide sequence heterogeneity of an RNA phage population Cell, 13 (4), 735-744
Domingo E. et al. (2012) Viral Quasispecies Evolution Microbiol. Mol. Biol. Rev. 2012, 76(2):159 - 216
Eigen M. (1971). Self-organization of matter and the evolution of biological macromolecules. Naturwissenschaften 58:465–523.
Eigen M., Schuster P. (1977). The hypercycle—a principle of natural self-organization. A. Emergence of the hypercycle. Naturwissenschaften 64:541–565.
Weissmann C. (1974). The making of a phage. FEBS Lett. 40(Suppl.):S10–S18.856.
Weissmann C. et al., 1973. Structure and function of phage RNA. Annu. Rev. Biochem. 42:303–328.
Anmerkung der Redaktion
Dieser Essay setzt eine Serie zum Thema virale Infektionen aus der Sicht der Theoretischen Biologie fort. Bisher sind erschienen:
Aus der Sicht des Biochemikers Gottfried Schatz: Spurensuche – wie der Kampf-gegen-Viren-unser-Erbgut-formte
Aus der Sicht des Virologen Peter Palese: Influenza Viren – Pandemien: sind universell wirksame Impfstoffe in Reichweite?
Weiterführende links
Siehe auch links zu den beiden oben genannten Essays
Zwei hervorragende, leicht verständliche (allerdings englische) Videos:
Introduction to Viruses and Viral Replication (Craig Savage)
http://www.youtube.com/watch?v=PEWjyx2TkM8&feature=endscreen 14:01min
Virology 2013 Lecture #22 – Evolution (Vincent Racaniello)
http://www.youtube.com/watch?v=2k3ZmuLlR9U 1:12:19
Vincent Racaniello ist ein sehr prominenter Virologe, Professor an der Columbia Universität, NY, der auch einen exzellenten blog betreibt, welcher zu den aktuellsten Problemen der Virologie Stellung nimmt: Virology blog http://www.virology.ws/
- Printer-friendly version
- Log in to post comments