Fr, 17.11.2011 - 04:20 — Inge Schuster
![]()

In der zentralen, hochromantischen Szene des Freischütz werden in der Wolfsschlucht Zauberkugeln gegossen, die nie ihr angepeiltes Ziel verfehlen. Die Treffsicherheit derartiger Zauberkugeln hat Paul Ehrlich, der Vater der Chemotherapie, vor rund hundert Jahren als Vision auf die erwünschte Wirkungsweise von Medikamenten übertragen.Ausgehend von den Versprechungen der damals noch jungen Chemie formulierte er: „Wir müssen chemisch zielen lernen“ und meinte damit, daß Medikamente spezifisch an den jeweils relevanten Krankheitserreger und nur an diesen andocken dürften. Dies bedeutete in den von ihm konkret behandelten Projekten: „Das Abtöten von Parasiten ohne erhebliche Schädigung des Organismus“.
Nach wie vor ist es oberstes Ziel der Pharmaforschung Arzneimittel zu finden, die bei einer angestrebten Wirkung frei sind von unerwünschten Nebeneffekten. Dies konnte aber weder zu Zeiten Ehrlichs verwirklicht werden (Ehrlich hatte mit Salvarsan zwar das erste wirksame Mittel gegen Syphilis gefunden, allerdings zeigte dieses erhebliche Nebenwirkungen) noch mit unseren heutigen Mitteln. Jeder Arzneistoff bindet im Organismus an eine Vielzahl von Strukturen und kann deren Eigenschaften modulieren. Das Spektrum der daraus resultierenden Nebenwirkungen reicht von harmlosen Effekten, manchmal auch durchaus positiven Begleiterscheinungen, bis hin zu schwer-und-schwerstwiegenden negativen Auswirkungen.
Die Weltgesundheits-Organisation (WHO) faßt dies in wenigen Sätzen zusammen [1]: “All medicines have side effects and some can be damaging. The effects of any treatment with a medicine cannot be predicted with absolute certainty. All medicines have both benefits and the potential for harm. People in every country of the world are affected by adverse drug reactions (ADRs). Unintended, harmful reactions to medicines – ADRs - are among the leading causes of death in many countries.”
Wie häufig treten schwere und schwerste Nebenwirkungen von Arzneimitteln (ADRs) auf?
Sehr umfangreiche Studien in den USA und England geben an, daß rund 6,6% aller Aufnahmen in Spitäler auf Grund von ADRs erfolgen [2]. Von den Patienten, die bereits wegen anderer Ursachen in stationärer Behandlung sind, erleiden rund 15% Nebenwirkungen, deretwegen sich der Spitalsaufenthalt bedeutend verlängern kann [3]. Vergleichbare Zahlen kommen aus vielen anderen Ländern, vergleichbar sind diese auch hinsichtlich schwerster, zum Tod führender Nebenwirkungen, die an 4.- 6. Stelle der Todesursachen rangieren [3, 4]. Berücksichtigt man, daß Nebenwirkungen bei Patienten in ambulanter Behandlung nicht ausreichend dokumentiert werden, dürfte das gesamte Problem noch wesentlich gravierender sein.
Welche Maßnahmen können getroffen werden um die für betroffene Patienten höchst schädigende, für Institutionen und natürlich auch Leistungsträger höchst unbefriedigende Situation zu verbessern?
Nach Meinung der WHO sollten zumindest 60% der ADRs vermeidbar sein. Deren Ursachen sind u.a:
- eine fehlerhafte Diagnose und damit die Verschreibung einer falschen Medizin oder falschen Dosis einer an und für sich richtigen Medizin,
- das Nichteinhalten der Anweisungen zur Anwendung eines Arzneimittels („Non-Compliance“),
- eine naive Selbstbehandlung mit Arzneimitteln und
- die individuelle genetische Prädisposition des Patienten [1].
Wenn man an diesem multifaktoriellen Rad in Richtung Reduktion von ADRs drehen möchte, kann dies – abgesehen von Fehldiagnosen und Schlampereien – durch bessere Information des Patienten (in einer ihm verständlichen Sprache) geschehen, mit dem Hinweis auf Nutzen und Risiken der empfohlenen Behandlung. Darüber hinaus sehe ich vor allem Chancen in der Berücksichtigung des individuellen genetischen Make-ups eines Patienten, welches Konzentration und Verweildauer eines Arzneimittels im Körper bestimmt und – damit z.T. zusammenhängend – in der Vermeidung von Mehrfach-Medikationen.
Was geschieht mit Fremdstoffen im Allgemeinen und Arzneistoffen im Besonderen im Organismus?
Unser Organismus, wie auch der aller höherer Lebewesen, ist permanent einer ungeheuren Vielfalt und Vielzahl an Fremdstoffen aus Umwelt und Nahrung ausgesetzt, die über Kontakt mit der Körperoberfläche, über das Verdauungssystem oder über die Atemwege in den Organismus gelangen können. In besonderem Maße sind kleine, fettlösliche (lipophile) Fremdstoffe geeignet sich in Fettgewebe und in den Lipid-Membranen, die Zellen und Organellen innerhalb der Zellen umschließen, einzulagern. Ohne einen entsprechenden Schutzmechanismus würde dies zu einer enormen Akkumulation derartiger lipophiler Verbindungen führen und daraus resultierend zum Versagen von Zell-und Organfunktionen.
Im Laufe der Evolution haben Organismen eine Reihe von Enzymen – Katalysatoren – entwickelt, die lipophile Fremdstoffe in zunehmend wasserlösliche Produkte (Metabolite) verwandeln können, welche eine geringere Akkumulation in Zellen zeigen und leichter aus dem Organismus ausgeschieden werden können (Abbildung 1).
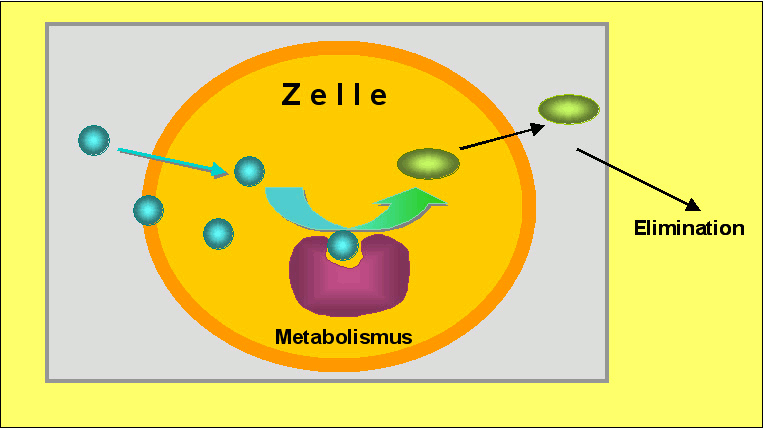 Abbildung 1. Fettlösliche (lipophile) Fremdstoffe (türkis), die in Zellen eintreten und sich darin anreichern, werden zu wasserlöslicheren, weniger akkumulierenden Metaboliten (grün) umgewandelt.
Abbildung 1. Fettlösliche (lipophile) Fremdstoffe (türkis), die in Zellen eintreten und sich darin anreichern, werden zu wasserlöslicheren, weniger akkumulierenden Metaboliten (grün) umgewandelt.
Im Prinzip ist dieses Set an Enzymen imstande, jedes bereits vorhandene und auch jedes in Zukunft noch zu synthetisierende Molekül zu Produkten umzuwandeln, die den Organismus über die üblichen Ausscheidungswege (Urin, Kot) verlassen können. (Nur wenige Typen von Molekülen, wie z.B. die Dioxine, leisten diesen Enzymen Widerstand, reichern daher bei fortgesetzter Exposition mehr und mehr im Organismus an und führen zu dessen massiver Schädigung).
Für die Fähigkeit verschiedenartigste Fremdstoffe metabolisieren zu können, bedarf es keiner sehr breiten Palette an unterschiedlichsten Enzymen, es genügt eine relativ kleine Gruppe, die sehr breite, zum Teil überlappende Spezifitäten für derartige Substanzen aufweisen. Eine besonders wichtige Rolle spielen dabei Enzyme aus der sogenannten Cytochrom P450 Familie (Eine Familie von Proteinen, die bereits in den primitivsten Lebensformen vorhanden waren und deren Struktur und Funktion über die Evolution konserviert wurde). Diese Enzyme (CYPs) sind für die Umwandlung von mehr als 75% aller lipophilen Fremdstoffe und damit auch für den Großteil an Arzneistoffen verantwortlich.
CYPs finden sich in praktisch allen Organen, besonders viel davon sitzt in Organen wie Leber und Darm, die über die Nahrungsaufnahme primär sehr hohen Konzentrationen an Fremdstoffen ausgesetzt sind. Für den Metabolismus der gebräuchlichen Arzneimittel sind - abhängig von deren jeweiligen Strukturen - nur wenige CYP-Formen verantwortlich (Abbildung 2): Der Großteil aller Pharmaka – vor allem relativ große Moleküle – werden von CYP3A4 umgewandelt, rund 20% von CYP2D6 und ein vergleichbarer Anteil von den nahe verwandten CYPs 2C9 und 2C19.
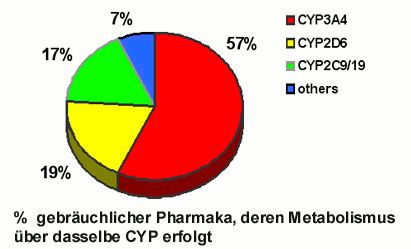 Abbildung 2. Nur wenige CYP-Formen sind für den Metabolismus aller gebräuchlichen Arzneistoffe verantwortlich.
Abbildung 2. Nur wenige CYP-Formen sind für den Metabolismus aller gebräuchlichen Arzneistoffe verantwortlich.
Konsequenzen der Metabolisierung von Arzneimitteln
Die Metabolisierung von Arzneistoffen führt zu einer Veränderung des Molekülcharakters und damit in den meisten Fällen zum raschen Verlust der erwünschten pharmakologischen Wirkung. Es hängt daher von der vorhandenen Menge und Aktivität der CYP-Formen ab, wie stark ein Medikament wirkt und wie lange seine Wirkung anhält.
Werden – wie es häufig der Fall ist – mehrere unterschiedliche Medikamente gleichzeitig angewandt, die über dieselbe CYP-Form metabolisiert werden, so konkurrrieren diese um dieses Enzym (sogenannte drug-drug interactions) und können sich gegenseitig in ihrem Abbau beeinträchtigen. Langsamerer Metabolismus führt zu längeranhaltenden höheren Konzentrationen und damit zu längerer Wirkung, steigert aber – entsprechend einer „Überdosierung“ – das Risiko für unerwünschte Nebenwirkungen.
Mehrfachmedikationen spielen eine besonders große Rolle bei Spitalspatienten (denen manchmal auch mehr als 15 unterschiedliche Medikamente pro Tag verabreicht werden) und ebenso auch bei Senioren aufgrund steigender Morbidität. Bei erhöhtem Substrat-Angebot können CYPs zwar verstärkt synthetisiert (induziert) werden, jedoch oft nicht ausreichend für Multi-Medikationen. Zunehmend demonstrieren Studien die klinischen Auswirkungen der drug-drug interactions, die besonders ausgeprägt sind, wenn es sich um die Konkurrenz für das CYP2D6 handelt. Dieses Enzym ist nur in relativ niedriger Konzentration vorhanden, aber für den Metabolismus von rund 20% aller gebräuchlichen Pharmaka verantwortlich (Abbildung 2), darunter befinden sich überaus wichtige, häufig gleichzeitig angewandte Klassen von Schmerzmitteln, Antidepressiva, Antipsychotika, Antiarrhythmika und Beta-Blockern.
Menschen unterscheiden sich in der Art und Weise, wie sie mit Arzneistoffen umgehen.
Ein besonderes Problem resultiert aus der genetischen Ausstattung einzelner Patienten. Gene der CYP-Enzyme weisen sogenannte Polymorphismen auf, d.h. es gibt Genvarianten die für CYP-Formen kodieren, deren Spektrum von nicht-funktionsfähigen über (normal-) funktionierende zu extrem rasch funktionierenden Enzymen reicht [5]. Je nachdem welche dieser Formen ein Mensch in seinem Genom geerbt hat, reagiert er in unterschiedlicher Weise auf Medikamente (und andere Fremdstoffe).
Von gravierender klinischer Bedeutung sind hier die Polymorphismen des oben bereits erwähnten CYP2D6: Bis zu 10% der Bevölkerung besitzen kein funktionsfähiges Enzym, d.h. sie können entsprechende Medikamente nicht oder nur sehr langsam (über andere Enzyme) abbauen, das Risiko für Nebenwirkungen ist damit hoch, insbesondere bei (der häufig entbehrlichen) Multi-medikation (siehe oben). Eine ebenso große Bevölkerungsgruppe besitzt ultra-rasch metabolisierende CYP2D6-Formen, d.h. entsprechende Medikamente werden zu rasch abgebaut und können teilweise überhaupt nicht ihre Wirkung entfalten. Was dies für die Therapie mit den oben angegebenen Medikamenten bedeutet, braucht wohl nicht näher angeführt werden.
Pharmakogenetische Tests zur Erhöhung der therapeutischen Wirkung und Vermeidung von Nebenwirkungen
Die junge Wissenschaft Pharmakogenetik untersucht den Einfluß des genetischen „Make-ups“ von Patienten auf Wirkung und Nebenwirkungen von Pharmaka. Als prominentes Beispiel für die erfolgreiche Anwendung pharmakogenetischer Methoden, wird in Fachliteratur und Populärliteratur die Testung auf Genvarianten von CYPs angeführt. Hier ist bereits seit mehreren Jahren ein Bio-Chip am Markt, der es erlaubt, auf die häufigsten Genvarianten von CYP2D6 (und auch CYP2C19) zu prüfen. Dieser Test ist mit rund 1000 € zwar nicht billig, erlaubt es aber einem großen Teil der Untersuchten festzustellen, welcher Gruppe von „Metabolisierern“ sie angehören. Mit dieser Information kann einerseits die Verschreibung von Medikamenten vermieden werden, die schlecht oder gar nicht metabolisiert würden und damit zu schweren Nebenwirkungen führen könnten. Andererseits kann hier und auch im Falle von ultra-rasch Metabolisierern durch entsprechende Adjustierung des Dosierungsschemas die erwünschte Wirkung bei gleichzeitig gemindertem Risiko für schwere Nebenwirkungen erzielt werden.
Die Übernahme der Kosten derartiger Testungen durch öffentliche Kassen sollte für unser Gesundheitssystem prioritäre Bedeutung haben: Patienten würden entscheidend profitieren, wenn sie nicht mehr mit der Wirkung schwerste Nebenwirkungen riskierten oder ein Medikament nach dem anderen auszuprobieren bräuchten, bis endlich eines wirkte. Es würden aber auch die Kosten für das Gesundheitssystem bedeutend sinken, infolge reduzierter Ausgaben für Medikamente ebenso wie infolge insgesamt reduzierter Behandlungskosten.
Einzelnachweise und Quellen:
[1] WHO (2008): http://www.who.int/mediacentre/factsheets/fs293/en/
[2] Pirmohamed M, et al (2004): Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. Bmj 329: 15–19
[3] Pirmohamed M. (2010) Drug-drug interactions and adverse drug reactions: separating the wheat from the chaff. Wien Klin Wochenschr 122:62-64
[4] Lazarou J, et al., (1998) Incidence of adverse drug reactions in hospitalized patients: a metaanalysis of prospective studies. Jama 279: 1200–1205
[5] Ingelman-Sundberg M, et al., (2007) Influence of cytochrome P450 polymorphisms on drug therapies: Pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther 116: 496-526
- Printer-friendly version
- Log in to post comments