Die Proteindatenbank: Strukturen, Modelle und zwingend erforderliche Korrekturen
Die Proteindatenbank: Strukturen, Modelle und zwingend erforderliche KorrekturenDo, 26.01.2017 - 08:03 — Bernhard Rupp & Inge Schuster 

![]()
Mit bis jetzt mehr als 120 000 eingespeisten Strukturdaten von Proteinen und daraus erstellten Modellen ist die Proteindatenbank (PDB) zur unentbehrlichen Basis von Grundlagenforschung und angewandter Forschung geworden. Allerdings kontaminieren einige wenige, stark fehlerhafte und sogar erfundene Strukturmodelle die Datenbank und beeinträchtigen Data-Mining, Metaanalysen und vor allem Wissenschafter die auf Basis der Daten aussichtlose, nur Ressourcen vergeudende Untersuchungen starten. Der aus Wien stammende Strukturbiologe Bernd Rupp ruft zu einem gemeinsamen Vorgehen von Strukturbiologen und Herausgebern von Fachzeitschriften auf, um derartige Einträge effizient zu eliminieren.*
Vor knapp 60 Jahren gelang einer der größten Durchbrüche in den Biowissenschaften: John Kendrew und Max Perutz veröffentlichten die ersten dreidimensionalen Strukturen von Proteinen. Es handelte sich dabei um die Strukturmodelle zweier verwandter, Sauerstoff bindender Proteine - Myoglobin und Hämoglobin, die in Jahrzehnte langer, überaus schwieriger Arbeit mittels Röntgenstrukturanalyse bestimmt worden waren. Die räumliche Lage der Atome zu einander war an Hand der Beugungsbilder von Röntgenstrahlen an den Proteinkristallen ermittelt worden. Man erkannte daraus nicht nur, wie sich Proteine falten, sondern konnte nun erstmals deren Funktion auf molekularer Basis verstehen: wie Hämoglobin den Sauerstoff in der Blutbahn aufnimmt, transportiert und abgibt, wie ein genetischer Defekt diese Vorgänge hemmt und wie Myoglobin in der Muskelzelle den Sauerstoff speichert.
Es dauerte dann bis 1967 bis die Struktur eines weiteren Proteins, des aus Hühnereiweiss isolierten Enzyms Lysozym, aufgeklärt wurde. In den nächsten Jahren folgten einige weitere Enzyme (u.a. Chymotrypsin, Carboanhydrase und Laktatdehydrogenase). Erstmals erkannte man nun die molekularen Details, wie Enzyme chemische Reaktionen katalysieren.
Die Protein Datenbank (PDB)
Eine kleine Tagung über "Struktur und Funktion von Proteinen auf 3D-Niveau" am Cold Spring Harbor Laboratory (NY, USA) brachte im Jahr 1971 die Pioniere der Strukturbiologie mit Biochemikern/Biophysikern zusammen, welche die Röntgen-Strukturdaten als Grundlage ihrer Forschung nutzen wollten. Man ahnte, dass dieses neue Gebiet eine Revolution für die gesamte Biologie darstellen würde und beschloss die Gründung einer zentralen Proteindatenbank PDB (untergebracht im Brookhaven National Laboratory, NY). In dieser sollten alle neuen Strukturdaten hinterlegt und für jeden Wissenschafter frei zugänglich sein. Damit begann der Siegeszug der Strukturbiologie. Wuchs diese Datenbank anfangs nur langsam - 1980 enthielt sie Daten von 69 Strukturen, 1990 von 507 Strukturen -, so ermöglichten rasche technologische Entwicklungen in Produktion, Reinigung und Kristallisation von Proteinen immer schnellere Analysen von immer komplizierter aufgebauten Systemen und zu deren Analyse und Visualisierung wurden immer bessere Verfahren entwickelt wurden. PDB wurde zu einer weltweiten Kollaboration (http://www.wwpdb.org/) von Datenbanken in den US (RSCB PDB), Japan (PDBj) und Europa (PDBe), in den letzten Jahren wurden jeweils mehr als 10 000 Strukturmodelle hinterlegt. Abbildung 1.
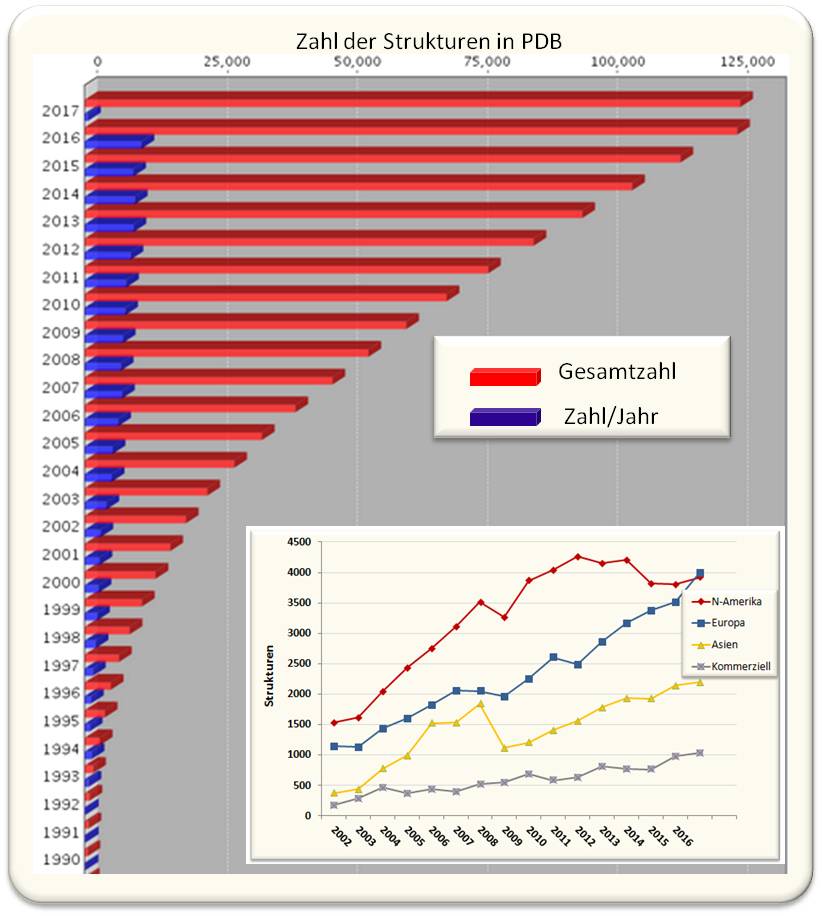 Abbildung 1. Zahl der in der Proteindatenbank (PDB) hinterlegten Strukturen. In den letzten Jahren sind jeweils rund 10 000 neue Strukturen hinterlegt worden. Das Insert unten zeigt, woher die Autoren dieser Daten stammten. Rund 10 % der Strukturen kommen aus der Industrie, insbesondere aus der pharmazeutischen Industrie, die so selektive Inhibitoren für Targetproteine - beispielsweise für die HIV-Protease - designt. (http://www.rcsb.org/pdb/statistics/contentGrowthChart.do?content=total&seqid=100,Update 17.01.2017)
Abbildung 1. Zahl der in der Proteindatenbank (PDB) hinterlegten Strukturen. In den letzten Jahren sind jeweils rund 10 000 neue Strukturen hinterlegt worden. Das Insert unten zeigt, woher die Autoren dieser Daten stammten. Rund 10 % der Strukturen kommen aus der Industrie, insbesondere aus der pharmazeutischen Industrie, die so selektive Inhibitoren für Targetproteine - beispielsweise für die HIV-Protease - designt. (http://www.rcsb.org/pdb/statistics/contentGrowthChart.do?content=total&seqid=100,Update 17.01.2017)
Es wurden und werden Strukturen von kleinsten bis zu enorm großen Proteinen aufgeklärt, allein und in Wechselwirkung mit ihren natürlichen Liganden (z.B. Hormonrezeptoren mit ihren Hormonen) und mit Molekülen die diese Interaktionen modulieren. Es liegen Analysen riesiger Proteinkomplexe (z.B. das Proteasom), ganzer Viren und Ribosomen vor. Als besonderes Highlight im Jahr 2016 ist hier die erste Struktur des Zika-Virus zu nennen, gefolgt von seinem Komplex mit einem Antikörper
Derzeit enthält die PDB insgesamt 126 060 Strukturmodelle, die zum überwiegenden Teil (92,8 %) von Proteinen und zum kleineren Teil auch von Nukleinsäuren stammen. Rund 90 % der Proteinstrukturen wurden durch Röntgenstrukturanalyse ermittelt, etwa 9 % durch Kernresonanzuntersuchungen(NMR).
Das Interesse an diesen mehr als 120 000 Strukturen ist enorm. Im vergangenen Jahr zählte die Website weltweit mehr als eine Million unterschiedlicher Besucher: Studenten und Forscher in biologischen, biomedizinisch/pharmazeutischen und ökologischen Fachrichtungen. Strukturdaten wurden rund 600 Millionen Mal abgerufen. ( http://cdn.rcsb.org/rcsb-pdb/general_information/news_publications/newsletters/2017q1/home.html).
Von Beugungsdaten zu Proteinmodellen
Liest man Veröffentlichungen, die auf der Analyse von makromolekularen Strukturen basieren, so erwartet man natürlich, dass die dazu gehörigen, in der PDB hinterlegten Strukturmodelle sachgerecht erstellt und verfeinert wurden. Die Strukturbiologen waren ja Vorreiter im Erstellen von Normen für die Hinterlegung von Daten und Modellen. Die meisten wissenschaftlichen Journale sind diesen ethischen Normen gefolgt und haben eine verpflichtende Hinterlegung der Modell Koordinaten und in jüngerer Zeit der Beugungsdaten gefordert.
Nun zeichnet sich der Weg von den rohen Beugungsdaten zu den prozessierten Strukturdaten (wie sie aktuell hinterlegt werden) und von hier zur Rekonstruktion der Elektronendichteverteilung durch große mathematischer Objektivität aus (vereinfacht dargestellt in Abbildung 2). Die Übersetzung der Elektronendichte in ein Atommodell erlaubt dagegen erheblichen Freiraum, der umso größer wird, je niedriger die Qualität der Beugungsmuster und das Fachwissen des Modellierers sind. Wenn man also erwartet, dass jedes Modell die in Form von Elektronendichte vorliegende Evidenz genau widerspiegelt, , ist dies gelegentlich eine arge (Ent)Täuschung.
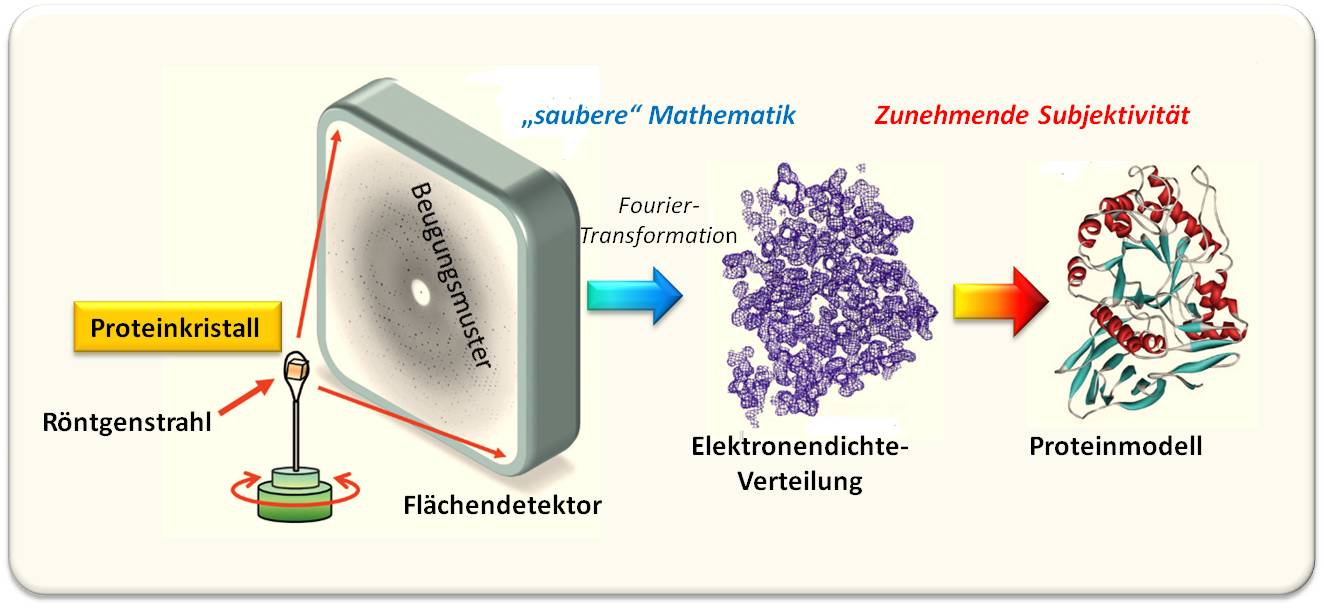 Abbildung 2. Röntgenkristallographie eines Proteins. Ein Proteinkristall wird fein-gebündelten (kollimierten), intensiven Röntgenstrahlen ausgesetzt, die an den Elektronenhüllen der Atome gebeugt werden. Die Beugungsbilder werden auf einem Flächendetektor aufgenommen und zu einem Set von Beugungsdaten zusammengesetzt – einem reziproken Code für die räumliche Anordnung der Atome im Kristall. Die Übersetzung diese Codes mittels Fourier-Transformation und Kenntnis der zugehörigen Phasenwinkel erlaubt die Rekonstruktion der Elektronendichte für die streuenden Atome. Daraus kann ein dreidimensionales Modell der Proteinstruktur erstellt werden - hier dargestellt in Form eines Ribbon – Modells (Abbildung modifiziert nach © Garland Science 2010).
Abbildung 2. Röntgenkristallographie eines Proteins. Ein Proteinkristall wird fein-gebündelten (kollimierten), intensiven Röntgenstrahlen ausgesetzt, die an den Elektronenhüllen der Atome gebeugt werden. Die Beugungsbilder werden auf einem Flächendetektor aufgenommen und zu einem Set von Beugungsdaten zusammengesetzt – einem reziproken Code für die räumliche Anordnung der Atome im Kristall. Die Übersetzung diese Codes mittels Fourier-Transformation und Kenntnis der zugehörigen Phasenwinkel erlaubt die Rekonstruktion der Elektronendichte für die streuenden Atome. Daraus kann ein dreidimensionales Modell der Proteinstruktur erstellt werden - hier dargestellt in Form eines Ribbon – Modells (Abbildung modifiziert nach © Garland Science 2010).
Falsche und unglaubwürdige Modelle
Praktisch alle Fachgesellschaften und Herausgeber von Fachzeitschriften begrüßten den Ruf der Strukturbiologen nach Hinterlegung von Beugungsdaten und Modellkoordinaten. Dagegen wurde der Frage, was man eigentlich mit Publikationen machen sollte, die nachgewiesenermaßen unrichtige Strukturmodelle enthielten und wie - um die Integrität öffentlicher Datenbanken zu erhalten - derartige Modelle dort gelöscht werden könnten, wesentlich weniger Aufmerksamkeit gezollt.
Falsche und unglaubwürdige Modelle sind kein belangloses Ärgernis. Sie erschweren die Extraktion von Daten (Data-Mining), beeinträchtigen Metaanalysen und beschädigen sicherlich auch Image und Glaubwürdigkeit von Journalen, die strukturbiologische Untersuchungen veröffentlichen. Überdies können Veröffentlichungen, die unrichtige Modelle enthalten, andere Wissenschafter zu aussichtslosen, bloß Ressourcen vergeudenden Versuchen verleiten - ein Dominoeffekt, der nur schwierig zu stoppen ist.
Es erscheint sehr wichtig, dass sich die Gemeinschaft der Strukturbiologen zusammen mit den Herausgebern von Journalen und den Begutachtern auf ein klar umrissenes Protokoll einigen, wie umgehend auf Veröffentlichungen reagiert werden sollte, die in ihren Strukturmodellen nachgewiesene schwere Fehlerenthalten und wie derartige Modelle in Datenbanken wirkungsvoll markiert oder gelöscht werden.
Zurückziehen (obsoleting) von Einträgen in der PDB
Gegenwärtig wird das Problem einer Kontamination der Datenbank durch die Politik der PDB verschärft, dass ein Zurückziehen ("obsoleting" in der Sprache der PDB) der Modell-Koordinaten nur möglich ist, wenn der Autor der hinterlegten Daten es verlangt oder erlaubt. Ein kritisierter Autor stimmt diesem Schritt aber nur selten zu. Insgesamt sind bis jetzt in der PDB von mehr als 120 000 Einträgen nur 3 557 zurückgezogen worden, jedoch häufig nur, weil die Autoren nun bessere experimentelle Daten oder verbesserte Modelle produziert hatten.
Einige wenige ausgewählte Beispiele aus den letzten zehn Jahren sollen einen Eindruck vermitteln, wie schwierig es ist Einträge in Fachjournalen und in der PDB zu korrigieren, sogar wenn es sich um nicht plausible Modelle oder gar nachgewiesene Fabrikationen handelt .
- Im Jahr 2006 erschien im Top-Journal Nature eine gefälschte Struktur des Komplement-Proteins C3b, das eine wichtige Rolle in der Aktivierung und Regulierung des Immunsystems zur Infektabwehr spielt. Ein kritischer Kommentar stellte die Fälschung sofort fest, wurde im Journal allerdings erst sechs Monate später online gestellt. Die Universität Alabama - Heimatuniversität des C3b-Kristallographen - untersuchte dann den Fall, stellte 2009 die Fälschung fest und, dass noch weitere 11 von dem Autor publizierte und in die PDB eingespeiste Strukturen verschiedener Proteine gefälscht waren. Hinsichtlich der C3b-Struktur meldete Nature erst 2016 das Zurückziehen der ursprünglichen Arbeit. Die Reaktion des Journals führte dann zur Löschung des Eintrags (Code 2HR0) in der PDB - obwohl der Autor nicht zustimmte. Von den anderen 11 inkriminierten Strukturen befinden sich aktuell noch 7 in der PDB (http://www.wwpdb.org/documentation/UAB)
- Ein weiteres Beispiel, wo zwischen Kritik und Rückziehen der Struktur enorm lange Zeit verstrich, liegt im Fall des 2000 publizierten Komplexes von Botulinumtoxin mit einer Targetsequenz (einem kurzen Peptid) des Proteins Synaptobrevin vor (Synaptobrevin ist in der neuronalen Signalübertragung essentiell und wird bereits durch kleinste Konzentrationen Botulinumtoxin inaktiviert). Obwohl auf Grund der Elektronendichteverteilung das Fehlen des Peptids bereits 2001 aufgezeigt wurde, wurde das Modell des angeblichen Komplexes erst 2009 zurückgezogen.
- Weitere Beispiele betreffen u.a. Modelle für Antikörper-Antigen Komplexe, die 2006 im Journal Immunity erschienen sind. Die Autoren stellen hier die Hypothese auf, dass das limitierte primäre Repertoire an Antikörpern dadurch erweitert wird, dass ein einzelner Antikörper jeweils verschiedene Antigene an unterschiedlichen Stellen binden kann. Vom Herausgeber in einem Leitartikel prominent herausgestellt, wurde bereits in 69 Publikationen auf diesen attraktiven Mechanismus Bezug genommen. Das Problem ist, dass sowohl die abwesende Elektronendichte als auch die praktisch unmögliche Stereochemie des Modelles nicht auf derartige Lokalisationen der Antigene im Komplex schließen lässt.
Damit ein fauler Apfel nicht das ganze Fass verdirbt
Stark fehlerhafte und sogar erfundene Strukturmodelle von Biomolekülen sind zwar selten, persistieren aber in den Datenbanken. Die sich daraus ergebenden Probleme - die Integrität der Datenbanken selbst und die Persistenz der auf falschen Strukturmodellen basierenden Arbeiten - müssen wirkungsvoll angegangen werden. Es ist ein Aufwand, der nicht nur kritischen Stimmen überlassen werden darf, die sich die Mühe machen die Irrtümer aufzuzeigen (und meistens ignoriert werden). Die gesamte Gemeinschaft der Strukturbiologen und in besonderem Maße die Herausgeber von Fachzeitschriften sind gefordert in einen konstruktiven Dialog einzutreten , damit die Strukturbiologie nicht ihre Glaubwürdigkeit als Evidenz-basierte Wissenschaft verliert.
* Ein ausführlicher Artikel von Bernhard Rupp und Kollegen über dieses Thema ist eben erschienen: B.Rupp et al., Correcting the record of structural publications requires joint effort of the community and journal editors. FEBS Journal 283 (2016) 4452–4457. doi:10.1111/febs.13765.
Weiterführende Links
Ein wunderschöner 2017 Kalender der PDB: http://pdb101.rcsb.org/learn/resource/2017-calendar-geis-digital-archive-calendar (RCSB PDB has published a 2017 calendar highlighting the work of Irving Geis (1908-1997) "Geis was a gifted artist who helped illuminate the field of structural biology with his iconic images of DNA, hemoglobin, and other important macromolecules")
Bernhard Rupp (2009): „Biomolecular Crystallography: Prinicples, Practice, and Application to Structural Biology“ (Garland Science, Taylor & Francis)- ein weltweit anerkanntes Lehrbuch.
Artikel über Kristallstrukturen im ScienceBlog
- Bernhard Rupp, 21.03.2014: Wunderwelt der Kristalle — Die Kristallographie feiert ihren 100. Geburtstag
- Bernhard Rupp, 04.04.2014: Wunderwelt der Kristalle — Von der Proteinstruktur zum Design neuer Therapeutika
- Gottfried Schatz, 17.01.2014: Porträt eines Proteins — Die Komplexität lebender Materie als Vermittlerin zwischen Wissenschaft und Kunst
- Patrick Cramer, 26.08.2016: Wie Gene aktiv werden.