Alzheimer-Forschung - richtungsweisende Studien dürften gefälscht sein
Alzheimer-Forschung - richtungsweisende Studien dürften gefälscht seinFr, 14.08.2022 — Inge Schuster

![]()
Eine 2006 erschienene Untersuchung, die erstmals über einen kausalen Zusammenhang zwischen dem Auftreten des Amyloid-Komplexes Ab*56 und dem kognitiven Abbau bei der Alzheimerkrankheit berichtete, erregte enormes Interesse, da sie eine massive Untermauerung der Amyloid-Hypothese zur Entstehung der Krankheit war. Der mehr als 3000 Mal zitierte Artikel wurde so richtungsweisend für die weitere, milliardenschwere Finanzierung zu Forschung und Entwicklung neuer Alzheimer-Therapeutika. Wie sich nun herausstellte, dürften die Forscher in dieser und auch in einer Reihe darauf folgender Arbeiten ihre Aussagen mit manipulierten Abbildungen belegt haben. Darüber hat das Fachjournal Science nach ausführlichen Recherchen Ende Juli 2022 einen langen, bestürzenden Bericht gebracht [1].
116 Jahre nachdem der Arzt Alois Alzheimer vor Irrenärzten in Tübingen einen Vortrag "Über eine eigenartige Erkrankung der Hirnrinde" hielt, in dem er sogenannte Plaques im Hirn einer Demenzkranken zeigte, stellt die nach ihm benannte Erkrankung ein noch immer ungelöstes Problem für die Weltgesundheit dar. Es ist weder klar, wodurch die Krankheit ausgelöst wird, noch gibt es Therapien, die den fortschreitenden kognitiven Abbau stoppen oder gar heilen könnten. Charakteristisch für die Alzheimer-Krankheit sind Proteinablagerungen die sich in Form von Plaques zwischen den Nervenzellen (β- Amyloid-Plaques) und als Knäuel von Fibrillen des Tau-Proteins im Innern der Nervenzellen bilden. Alzheimer betrifft vor allem alte Menschen, kann aber auch schon in jüngerem Alter, d.i. unter 65 Jahren, auftreten; das zumeist langsame, sich über Jahre erstreckende Absterben von Nervenzellen ist für den überwiegenden Teil (bis zu 70 %) aller Demenzerkrankungen verantwortlich.
WHO hat Demenz zur Priorität für die Weltgesundheit erklärt
https://www.who.int/news-room/fact-sheets/detail/dementia
Laut WHO sind global derzeit rund 55 Millionen Menschen von Demenz - und dies bedeutet überwiegend von Alzheimer - betroffen, rund 10 Millionen Erkrankte kommen jährlich hinzu; mit der Zunahme der Bevölkerung und den noch schneller wachsenden älteren Gruppen wird die Zahl der Kranken im Jahr 2030 voraussichtlich auf 78 Millionen und im Jahr 2050 auf 139 Millionen (das sind dann rund 1,4 % der Weltbevölkerung) ansteigen. Demenz ist weltweit die siebenhäufigste Todesursache und einer der Hauptgründe für Behinderungen und Pflegebedürftigkeit von älteren Menschen. Die Erkrankung hat physische, psychische, soziale und wirtschaftliche Auswirkungen nicht nur für die Betroffenen, sondern auch für ihre Betreuer, Familien und die Gesellschaft insgesamt. Wurden die weltweiten gesellschaftlichen Gesamtkosten von Demenz im Jahr 2019 auf 1,3 Billionen US-Dollar geschätzt, so wird bis 2030 ein Anstieg auf 2,8 Billionen US-Dollar erwartet. Mit dem Global action plan on the public health response to dementia 2017-2025 ein umfassendes Handlungskonzept für politische Entscheidungsträger, internationale, regionale und nationale Partner und die WHO ins Leben gerufen worden.
Fehlende Therapeutika
Die einzigen zugelassenen Medikamente gegen Alzheimer können die Neurodegeneration nicht stoppen, sondern lediglich die Symptome behandeln - dies nicht sehr effizient und mit zum Teil schweren Nebenwirkungen.
Insgesamt wurden bis jetzt 7 Pharmaka zugelassen, 5 davon sind auf Neurotransmitter abzielende Wirkstoffe, die schon vor mehr als 25 Jahren auf den Markt kamen: Inhibitoren der Acetylcholinesterase (Donezepil, Galantamin, Rivastigmin und Tacrin) und ein NMDA-Rezeptor Antagonist (Memantin). Suvorexant, ein Antagonist des Orexin-Rezeptors wurde eigentlich zur Behandlung von Schlaflosigkeit zugelassen, von der häufig auch Alzheimer-Kranke betroffen sind. Basierend auf der Hypothese, dass eine Verringerung der zwischen den Nervenzellen abgelagerten Plaques ein Fortschreiten der Erkrankung aufhalten/umkehren könnte (s.u.), wurden 1984 als deren Beta-Amyloid-Peptide identifizierte Komponenten rasch zu wichtigen Zielstrukturen (Targets) der Alzheimer-Forschung und Entwicklung. Allerdings sind die Bemühungen jahrzehntelang an fehlender Wirksamkeit und/oder nicht tolerablen Nebenwirkungen gescheitert. Dass 2021 mit Aduhelm (Aducanumab) erstmals ein gegen β-Amyloid-Proteine gerichteter Antikörper, von der FDA zugelassen wurde, stellt wahrscheinlich keinen echten Durchbruch dar. Diese Zulassung ist bei führenden Experten auf heftige Ablehnung gestoßen, da Aduhelm zwar die Amyloid-Plaques reduzierte, dies aber nicht mit verbesserten kognitiven Fähigkeiten der Patienten korrelierte und dazu eine Reihe schwerer Nebenwirkungen auftraten. Die europäische Arzneimittelagentur EMA hat sich gegen eine Zulassung ausgesprochen.
Von insgesamt 331 gelisteten klinischen Alzheimer-Studien (klinische Phasen 1 - 3) gibt es 69, deren Target Beta-Amyloid ist - in den besonders großen klinischen Studien der entscheidenden Phase 3 sind es 9 von 23. Wie oben erwähnt sind viele der Amyloid-Studien bereits gescheitert, 30 wurden schon abgebrochen. (Eine detaillierte Auflistung verschiedener Targets in den einzelnen klinischen Phasen Liste findet sich in [2].)
Die Amyloid-Hypothese
geht von einer zentralen Rolle der Beta-Amyloid-Peptide in der Alzheimer-Erkrankung aus. Diese bestehen zumeist aus 38- 42 Aminosäuren langen Peptidketten, die mit Hilfe von Enzymen (Sekretasen) aus dem Vorläufer-Protein Amyloid-Precursor -Protein (APP) abgespalten werden. APP ist in vielen Körperzellen, insbesondere an den Synapsen der Nervenzellen exprimiert. Im gesunden Hirn dürften APP und auch seine Spaltprodukte wichtige Funktionen in physiologischen Prozessen spielen, u.a. in der Bildung von Synapsen und in der Neuroprotektion.
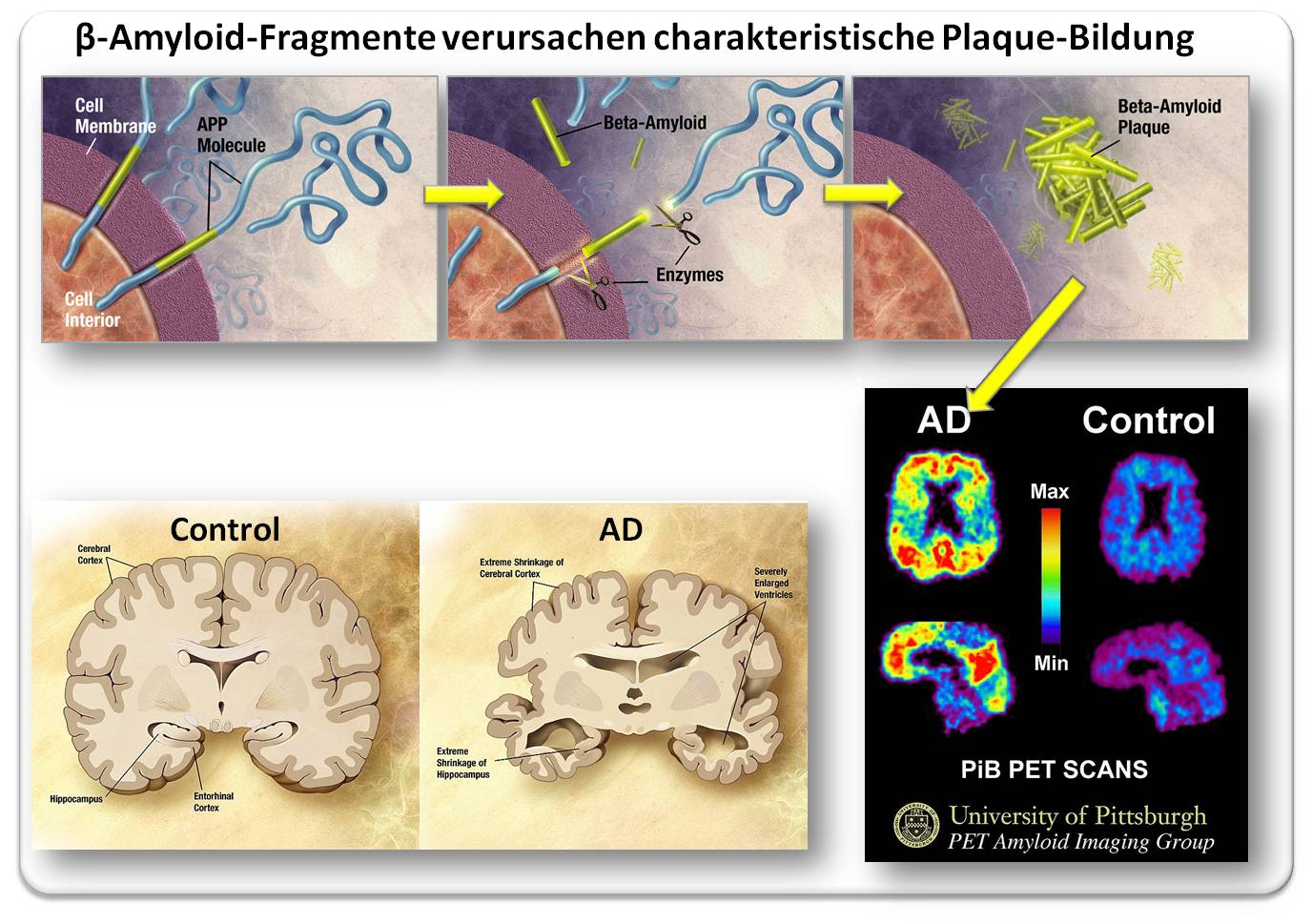
APP ist ein in der Zellmembran sitzendes Rezeptor-Protein, das mit dem Großteil seiner Sequenz aus der Zelle herausragt (Abbildung 1 oben). Die Abspaltung des langen, extrazellulären Teils (blau) der Kette führt u.a. zu kleinen, etwa 40 Aminosäuren großen, Bruchstücken (gelb), den sogenannten Beta-Amyloid-Peptiden. Diese können aggregieren und - wenn sie durch das glymphatische System des Gehirns nicht in ausreichendem Maße entsorgt und/oder abgebaut werden - zu unlöslichen Plaques zusammenwachsen (Abbildung 1 oben, rechts), die in weiterer Folge Entzündungen und Schädigungen von Nervenzellen und ihren Funktionen hervorrufen.
Durch geeignete Marker (in diesem Fall PiB) können solche Plaques nicht-invasiv mittels Positron-Emission-Tomographie (PET) sichtbar gemacht werden und damit die Alzheimer-Diagnose (AD) erhärten. Abbildung 1 (unten rechts) zeigt im Hirn eines Alzheimer-Kranken hohe Anreicherungen des Markers - und damit der Amyloid-Plaques - im Frontal- und Scheitellappen, Regionen, die für kognitive Prozesse, motorische Steuerung, operative Funktionen und Sinneswahrnehmungen verantwortlich sind. Dass solche massiven Ansammlungen schwere Beeinträchtigungen der lokalen Hirnfunktionen zur Folge haben sollten, erscheint einleuchtend.
Die Unterbindung von Nervenverbindungen und das massive Absterben von Nervenzellen führen bei fortschreitender Krankheit zu einer extremen Schrumpfung von Teilen des Gehirns insbesondere der Hirnrinde (Cortex) und des Hippocampus und zu hochgradig erweiterten Ventrikeln (Abbildung 1. links unten).
|
|
|
Abbildung 1: Von der Bildung der Beta-Amyloid-Peptide über die massive Anhäufung von Amyloid-Plaques in essentiellen Gehirnregionen von Alzheimer-Kranken (AD, sichtbar gemacht durch Positronen Emissions-Tomographie - PET) zur Gehirnschrumpfung. Erklärung: siehe Text. (Bilder oben aus http://www.nia.nih.gov/Alzheimers/Publications/UnravelingTheMystery/Part1/Hallmarks.htm ; Bild unten rechts: Klunkwe https://commons.wikimedia.org/wiki/File:PiB_PET_Images_AD.jpg, Lizenz: cc-by-sa; Bild unten links: gemeinfrei). |
Im Laufe der Jahre kam allerdings die Abfolge "Generierung von Beta-Amyloid - Bildung von Plaques - Demenz" ins Wanken, da sich herausstellte, dass die Plaque-Belastung nicht mit dem Erscheinungsbild der Krankheit korrelierte, Nervenzellen auch an Orten ohne Plaques abstarben, es anderseits nicht-demente Personen mit ausgeprägten Plaques gab und in klinischen Studien die Reduktion der Plaques keinen Einfluss auf das Krankheitsgeschehen hatte. In der Folge kam es zu einem Paradigmenwechsel: Untersuchungen deuteten nun darauf hin, dass nicht Amyloid-Plaques sondern kleinere lösliche Aggregate - Beta-Amyloid Oligomere - die eigentlichen toxischen Strukturen sein könnten.
Wäre nun vielleicht ein Amyloid-Oligomeres Auslöser der Alzheimer-Krankheit und damit endlich ein erfolgversprechendes Target für das Design wirksamer Therapeutika in Sicht?
Ein neues, toxisches Amyloid-Oligomer
Ein 2006 im Fachjournal Nature erschienener Artikel bestätigte die Hypothese vom toxischen Amyloid-Oligomer [3]. Forscher am Department of Neuroscience der Universität von Minnesota (Minneapolis) hatten an einem validierten Alzheimer-Mäusemodell (s.u.) ein neues Amyloid-Oligomeres, Ab*56, im Hirngewebe identifiziert, dessen Menge mit dem nachlassenden Gedächtnis der alternden Mäuse korrelierte (dies wurde an Hand des von den Tieren zuvor erlernten Orientierungsvermögens im sogenannten Morris Wasser-Labyrinth untersucht). Isoliert und in gereinigter Form in junge, gesunde Ratten injiziert löste Ab*56 in diesen Tieren dann Gedächtnisverlust aus. Damit schien erstmals ein kausaler Zusammenhang zwischen dem toxischen Amyloid-Oligomer und dem Gedächtnisverlust, also eine Hauptursache für die Erkrankung gefunden worden zu sein. "Unsere Daten zeigen, dass Ab*56 das Gedächtnis gesunder, junger Ratten beeinträchtigt, und stützen die Hypothese, dass Ab*56 die Hauptursache für den Gedächtnisverlust bei Tg2576-Mäusen mittleren Alters ist." schrieben die Forscher. Ihre Daten stützten sich auf eine Reihe aussagekräftiger Fotos von sogenannten Western-Blots, die Ab*56 und seinen Verlauf im Alterungsprozess des Gehirns dokumentierten. (nb: In Western Blots werden Proteinlösungen, die mittels Gelelektrophorese im elektrischen Feld nach Größe getrennt wurden, auf eine Membran übertragen (Blotting) und die einzelnen Proteinbanden mit immunologischen Methoden nachgewiesen.)Ab*56 wurde dann auch in menschlichen Gehirnen identifiziert, sein Gehalt stieg mit dem Alter an und korrelierte im frühen Stadium der Alzheimer-Krankheit mit der Plaque-Last und dem pathologischen Tau-Protein.
Die 2006 Studie löste ein enormes Echo aus - wurde seitdem zur meistzitierten Alzheimer-Arbeit (laut Google Scholar kam es zu 3276 Zitierungen) - und gab der bis dato erfolglosen, bereits stark kritisierten Amyloid-Forschung neuen Aufwind. Die amyloid-bezogene Alzheimer-Forschung erhielt enorme Unterstützung - fast die Hälfte der gesamten Alzheimer-Gelder sollen in die Amyloid-Forschung geflossen sein. Die US-National Institutes of Health (NIH) haben allein im letzten Jahr 1,6 Milliarden US Dollars - die Hälfte ihres Alzheimer Budgets - an Amyloid-bezogene Projekte vergeben - zu Lasten von Forschern, die andere Ideen entwickelten. Natürlich wechselten viele Alzheimer-Forscher auf das erfolgversprechende, finanziell besser unterstützte neue Thema.
Es gab damals wenig Grund an den Ergebnissen zu zweifeln, handelte es sich bei den Autoren offensichtlich doch um erfahrene, brillante Experten:
Sylvain Lesné,ein junger französischer Wissenschaftler, der Erstautor der Studie und Entdecker des toxischen Ab-Oligomer, hatte bereits in seiner Doktorarbeit in Frankreich über den Metabolismus des Amyloid-Precursor-Proteins gearbeitet. Unmittelbar danach war er 2002 als PostDoc zur hochrenommierten Neurobiologin Karen Ashe, Professorin am Neuroscience Department in Minneapolis gekommen. Ashe ist Koautorin der Studie [3] und auch einiger späterer Studien von Lesne. Sie gilt als Pionierin der Alzheimerforschung. Als PostDoc hatte sie im Labor von Stanley Pruisiner wesentlich zu dessen Prionen-Forschung beigetragen (für die er mit dem Nobelpreis ausgezeichnet wurde); an der University of Minnesota forscht sie nun seit 30 Jahren am Gedächtnisverlust bei der Alzheimer-Krankheit und hat u.a. ein Alzheimer-Mausmodell - eine transgene Maus entwickelt, die menschliches Beta-Amyloid produziert, welches im Gehirn des Tieres Plaques bildet - das Modell wird weltweit angewandt.
In den folgenden 16 Jahren sind eine Reihe weiterer Publikationen von Lesné und Ashe - gemeinsam und auch einzeln - erschienen, welche die Befunde von 2006 bekräftigten und darauf aufbauten. Lesné wurde Assistant Professor, erhielt ein eigenes Labor und für seine Projekte insgesamt rund 10 Millionen US-Dollar Unterstützung von den NIH.
Merkwürdigerweise wurde - trotz des enormen Interesses an dem neuen Konzept - das toxische Ab*56 Amyloid von anderen Forschergruppen kaum nachgewiesen.
Ab*56 - Fact or Fiction...............
Im vergangenen Jahr ist der Neurowissenschaftler und Alzheimer-Forscher Matthew Schrag (Vanderbilt University) bei einer Recherche auf der PubPeer Website auf Kritik an den Lesné-Artikeln gestoßen, welche die Echtheit der Abbildungen zu Identifikation und Charakterisierung von Ab*56 in Frage stellten. Mit Hilfe von Software-Tools hat Schrag den 2006-Artikel und darauf folgende Artikel untersucht und in rund 20 davon mehr als 70 fragwürdige, manipulierte Western-Blots gefunden, die duplizierte, hineinkopierte Banden und Hintergründe aufwiesen [1].
Schrag hat seine Ergebnisse den NIH und den Fachjournalen Science und Nature gemeldet. Science beauftragte daraufhin unabhängige Bildanalytiker und mehrere führende Alzheimerforscher mit der Überprüfung von Schrags Ergebnissen zu den Artikeln von Lesné. Diese stimmten mit den Schlussfolgerungen von Schrag überein. "Die Bilder scheinen durch Zusammensetzen von Teilen von Fotos aus verschiedenen Experimenten zusammengestellt worden sein", sagte eine Expertin.[1]
Es sieht also derzeit so aus, als ob zahlreiche Abbildungen, die die Existenz und Funktion des Amyloid Oligomeren beweisen sollten, massiv manipuliert worden sind. Nach einem halben Jahr intensiver Recherche hat Science am 21. Juli 2022 über diesen Fall in einem langen ausführlichen Artikel berichtet.[1]
...........und Konsequenzen
Wie groß der durch diese Studien angerichtete Schaden für die Alzheimer Forschung aber auch ganz allgemein für das Vertrauen in die Wissenschaft ist, kann noch nicht abgeschätzt werden.
Gibt es das Amyloid-Oligomere Ab*56 überhaupt? Oder sind die publizierten Bilder vielleicht "nur" geschönt und das Amyloid in den Originalen vielleicht doch, wenn auch nicht so klar, nachgewiesen? Allerdings: Warum konnten es dann andere Forscher nicht finden? Lesné ist laut Science derzeit für eine Stellungnahme nicht erreichbar.
Warum sind die manipulierten Abbildungen Karen Ashe nicht aufgefallen, die ja Koautorin und anfangs Betreuerin ihres Postdocs Lesné war? Warum hat sie als Koautorin nicht in die Originalfotos Einsicht genommen? Auch als Alleinautorin eines Übersichtsartikels im Jahr 2010 bekräftigt sie in einem langen Kapitel die Ergebnisse aus dem 2006-Artikel und übernimmt einige der dortigen Abbildungen [4].
Warum sind die Abbildungen den Gutachtern der Publikationen nicht aufgefallen? Insbesondere im Journal of Neuroscience wurden fünf verdächtige Arbeiten veröffentlicht.
Was bedeutet dies aber nun für die Alzheimer-Forschung? Wurde diese durch die Befunde von Lesné 16 Jahre lang in die falsche Richtung gelenkt?
Der Direktors des National Institutes of Aging der NIH bemüht sich in einer "Erklärung zur Demenzforschung mit Amyloid-Beta-Protein" um Schadensbegrenzung [5]:
"Unter den identifizierten Oligomeren befindet sich eines mit der Bezeichnung Aβ*56. Während diese Entdeckung anfänglich ein gewisses Interesse weckte, führte sie zu einer begrenzten Reihe von nachfolgenden Forschungen, da es an spezifischen Markern fehlte, um es im Labor nachzuweisen, und da die anfänglichen Ergebnisse nicht reproduziert werden konnten. Es ist bemerkenswert, dass das Ab*56-Oligomer eines von vielen war, die damals erforscht wurden, und dass seitdem kein Alzheimer-Biomarker oder eine experimentelle Therapie auf der Grundlage von Ab*56 entwickelt wurde. Stattdessen stehen Immuntherapien, die auf Ab-Monomere (eine einzelne "Einheit" von Ab), andere Arten von Oligomeren und die längeren Amyloidfibrillen abzielen, im Mittelpunkt von Studien über potenzielle Medikamente zur wirksamen Behandlung von Demenz."
Deutlicher äußert sich der Nobelpreisträger Thomas Südhof, der selbst über Ursachen neuronaler Erkrankungen forscht [1]: "Der unmittelbare, offensichtliche Schaden ist die Verschwendung von NIH-Mitteln und die Verschwendung von Ideen in diesem Gebiet, weil die Leute solche Ergebnisse als Ausgangspunkt für ihre eigenen Experimente verwenden."
[1]Ch. Piller, Blots on a field? 21.7.2022. Science 377(6604):358-363. 10.1126/science.add9993
[2] Alzforum:https://www.alzforum.org/therapeutics
[3] Sylvain Lesne et al., A specific amyloid-b protein assembly in the brain impairs memory. Nature 440|16 March 2006|doi:10.1038/nature04533
[4] Karen H. Ashe, Animal models to study the biology of amyloid-beta protein misfolding in Alzheimer disease; in Protein Misfolding Diseases: Current and Emerging Principles and Therapies (Marina Ramirez-Alvarado et al., John Wiley & Sons, 2010) rb.gy/tez5zz
[5] Richard J. Hodes, M.D., Director, National Institute on Aging : NIA statement on amyloid beta protein dementia research. https://www.nia.nih.gov/news/nia-statement-amyloid-beta-protein-dementia-research
Weiterführende Links
Sylvain Lesné: homepage https://lesnelab.org/
Karen H. Ashe: homepage https://www.memory.umn.edu/research/ashe-lab/
National Institutes of Health (NIH). https://www.nih.gov/
Alzheimer: Eine dreidimensionale Entdeckungsreise. https://www.youtube.com/watch?v=paquj8hSdpc
Tau-Protein gegen Gedächtnisverlust (ohne Ton). Max-Planck Film 1:44 min, http://www.mpg.de/4282188/Tau-Protein_gegen_Gedaechtnisverlust
-----------------------------------------------
Artikel über die Alzheimer-Krankheit im ScienceBlog:
Francis S. Collins, 14.02.2019: Schlaflosigkeit fördert die Ausbreitung von toxischem Alzheimer-Protein.
Francis S. Collins, 27.05.2016: Die Alzheimerkrankheit: Tau-Protein zur frühen Prognose des Gedächtnisverlusts
Inge Schuster, 24.06.2016; Ein Dach mit 36 Löchern abdichten - vorsichtiger Optimismus in der Alzheimertherapie.
Gottfried Schatz, 03.07.2015:; Die bedrohliche Alzheimerkrankheit — Abschied vom Ich.